FARMACOCINÉTICA Y FARMACODINAMIA Dinámica de la absorción, distribución, acción y eliminación de los fármacos
La farmacoterapia efi caz requiere varios factores además de
una acción farmacológica conocida sobre un tejido específico
en un receptor particular. Cuando un fármaco penetra en el
organismo, de inmediato el cuerpo empieza a trabajar sobre
el mismo: lo absorbe, distribuye, metaboliza (biotransforma)
y elimina. Éstos son los pasos de la farmacocinética. Además,
el fármaco actúa en el organismo, interacción para la que es
esencial el concepto de un receptor farmacológico, puesto
que este receptor es el autor de la selectividad de la acción
farmacológica y de la relación cuantitativa entre el fármaco
y su efecto. Los mecanismos de la acción farmacológica son
los pasos de la farmacodinamia. El desarrollo cronológico de
la acción farmacológica terapéutica en el organismo puede
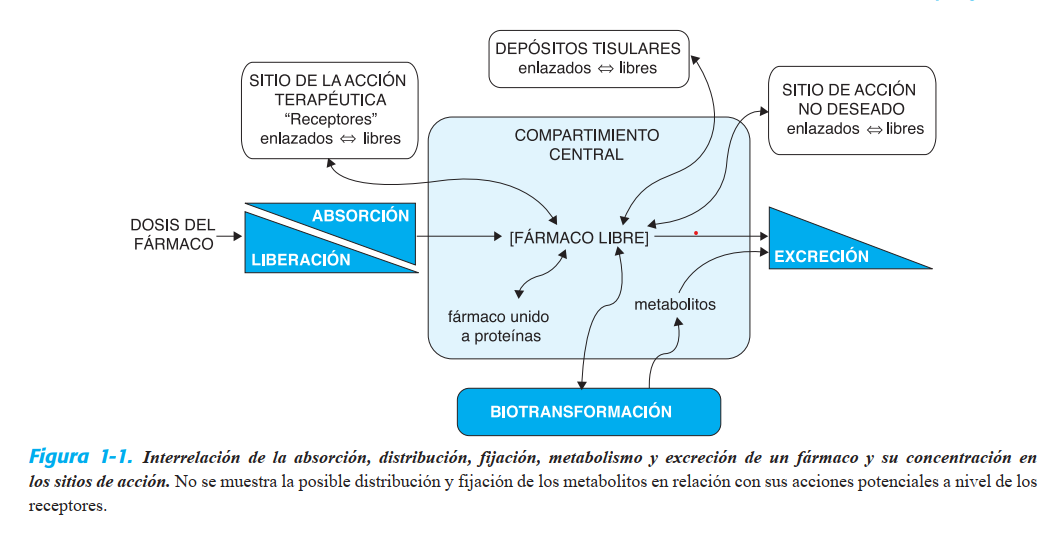
comprenderse en términos de farmacocinética y farmacodinamia (fig. 1-1)
I. FARMACOCINÉTICA: DINÁMICA DE LA ABSORCIÓN, DISTRIBUCIÓN, METABOLISMO Y EXCRECIÓN DE LOS FÁRMACOS
FACTORES FISICOQUÍMICOS EN LA TRANSFERENCIA DE LOS FÁRMACOS A TRAVÉS DE LAS MEMBRANAS
La absorción, distribución, metabolismo y excreción de un
fármaco suponen su paso a través de las membranas celulares. Es indispensable conocer los mecanismos por medio
de los cuales los fármacos las atraviesan, así como las propiedades fi sicoquímicas de las moléculas y las
membranas que modifican esta transferencia, para comprender la disposición de los fármacos en el cuerpo humano. Las características de
un fármaco que permiten pronosticar su desplazamiento y disponibilidad en los sitios de acción son su tamaño y forma
moleculares, grado de ionización, solubilidad relativa en lípidos de sus variantes ionizada y no ionizada y su enlace con
las proteínas séricas y tisulares (hísticas).
En la mayor parte de los casos, el fármaco debe atravesar las membranas plasmáticas de varias células para alcanzar su
sitio de acción. Si bien las barreras para el desplazamiento de los fármacos pueden ser una sola capa de células (epitelio
intestinal) o varias capas de células y proteínas extracelulares adjuntas (piel), la membrana plasmática representa la barrera
más común que deben atravesar los fármacos.
Membranas celulares. La membrana plasmática está formada por una
doble capa de lípidos anfi páticos, con sus cadenas de carbohidratos
orientadas hacia el interior para formar una fase hidrófoba continua, y
sus “cabezas” hidrófi las orientadas al exterior. Las moléculas de lípidos
individuales en la doble capa varían con la membrana en particular, y se
pueden mover en sentido lateral y organizarse con colesterol (p. ej., esfingolípidos), y así dar a la membrana propiedades como fluidez, fl exibilidad, gran resistencia eléctrica e impermeabilidad relativa a moléculas
fuertemente polares. Las proteínas de la membrana que están dentro de
la capa doble sirven como receptores, canales de iones, o transportadores que transducen vías de señalización eléctricas y químicas y constituyen blancos selectivos para la acción de medicamentos. Estas proteínas
algunas veces pueden asociarse a la caveolina y son secuestradas en
caveolae, otras veces son expulsadas de las caveolae y otras más se organizan en dominios de señales con abundante colesterol y esfingolípido
que no contiene caveolina
Las membranas celulares son relativamente permeables al agua, sea
por difusión o por la corriente que originan las diferencias hidrostáticas
u osmóticas a través de la membrana, y de la magnitud del fl ujo de agua
que puede arrastrar consigo moléculas de fármacos. Sin embargo, las moléculas de fármacos unidos a proteínas son demasiado grandes y polares
para este tipo de transporte. Por consiguiente, el desplazamiento a través
de la membrana por lo general se limita a los fármacos que se encuentran
libres. El transporte paracelular cruzando los espacios intercelulares es
suficiente como para que el desplazamiento a través de la mayor parte de
los capilares quede limitado por la circulación sanguínea y no por otros
factores (véase más adelante en este capítulo). Como se describe después,
este tipo de transporte es un factor importante en la fi ltración a través de
las membranas glomerulares renales. Sin embargo, existen algunas excepciones importantes a tal tipo de difusión capilar, puesto que ciertos
tejidos poseen uniones intercelulares “estrechas” y en ellas el transporte
paracelular es muy limitado. Los capilares del sistema nervioso central
(SNC) y de una gran variedad de tejidos epiteliales poseen uniones estrechas (véase más adelante en este capítulo). El fl ujo importante (masivo)
de agua puede llevar consigo sustancias hidrosolubles pequeñas, pero el
transporte del fl ujo a granel se limita cuando la masa molecular del soluto
es mayor de 100 a 200 daltones. Por lo tanto, la mayor parte de los fármacos lipófi los grandes deben atravesar la membrana celular misma.
Transporte pasivo a través de la membrana. Los fármacos atraviesan
las membranas ya sea por transporte pasivo o por mecanismos que comprenden la participación activa de ciertos componentes de la membrana.
En el transporte pasivo, la molécula de fármaco penetra por difusión a lo
largo de un gradiente de concentración gracias a su solubilidad en la capa
doble de lípidos. Este tipo de transferencia es directamente proporcional
a la magnitud del gradiente de concentración a través de la membrana,
al coeficiente de reparto entre lípidos y agua del fármaco y a la superficie de la membrana que tiene contacto con el fármaco. Entre mayor es
el coeficiente de reparto, mayor será la concentración de fármaco en la
membrana y más rápida su difusión. Una vez que se alcanza un estado
o condición estable, la concentración del fármaco libre es la misma en
ambos lados de la membrana, siempre y cuando el fármaco no sea un
electrólito. Para los compuestos iónicos, las concentraciones estables dependen del gradiente electroquímico para el ion y de las diferencias en el
pH a través de la membrana, que modifican el estado de ionización de la
molécula de manera desigual en ambos lados de la membrana.
Electrólitos débiles e influencia del pH. Casi todos los fármacos son ácidos o bases débiles que están en solución, en sus
formas ionizada o no ionizada. Las moléculas no ionizadas por
lo regular son liposolubles y se difunden a través de la membrana celular. En cambio, las moléculas ionizadas no pueden penetrar por la membrana lipídica, por su escasa liposolubilidad.
Por consiguiente, la distribución transmembrana de un
electrólito débil suele depender de su pKa y del gradiente de
pH entre uno y otro lados de la membrana. El pKa es el pH en
el cual la mitad del fármaco (electrólitos débiles) se halla en su
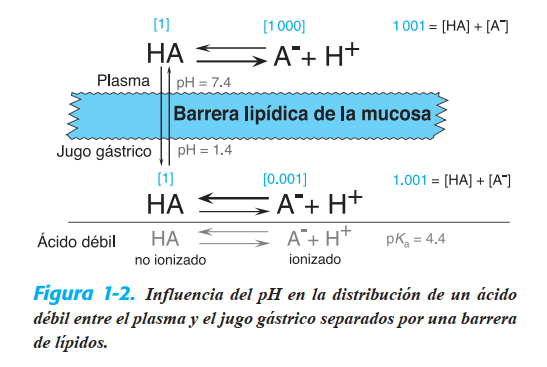
forma ionizada. Para ilustrar el efecto del pH en la distribución
de los medicamentos, en la figura 1-2 se muestra la partición o “reparto” de un ácido débil (pKa 5 4.4) entre el plasma
(pH 5 7.4) y el jugo gástrico (pH 5 1.4). Se supone que la mucosa gástrica se comporta como una barrera de lípidos simple,
que es permeable sólo a la forma liposoluble no ionizada de
la sustancia ácida. La razón aritmética entre las formas no ionizada e ionizada en cada valor de pH se calcula fácilmente
por medio de la ecuación de Henderson-Hasselbalch:
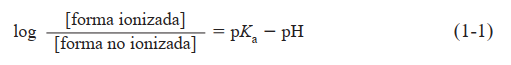
Esta ecuación correlaciona al pH del medio que rodea al
fármaco y a la constante de disociación ácida del fármaco
(pKa) con la razón entre las formas protonada (HA o BH1)
y no protonada (A2 o B), donde HA ↔ A2 1 H1 (Ka 5
[A2][H1]/[HA]) describe la disociación de un ácido y BH1
↔ B 1 H1 (Ka 5 [B][H1]/[BH1]) describe la disociación
de la forma protonada de una base.
De esta manera, en el plasma la razón de fármaco no ionizado a
medicamento ionizado es de 1:1 000; en el jugo gástrico, de 1:0.001.
Estos valores se señalan entre corchetes en la fi gura 1-2. Calculada
del mismo modo, la razón de la concentración total entre el plasma y el jugo gástrico sería de 1 000:1, si dicho sistema alcanzara
un estado estable. En el caso de una base débil con pKa de 4.4 (por
ejemplo, clorodiazepóxido), la razón se invertiría, al igual que las
flechas horizontales gruesas de la fi gura 1-2, que señalan la especie
predominante con cada valor de pH. Sobre tal base, en el estado
estable un fármaco ácido se acumulará en el lado más “alcalino”
de la membrana, y un fármaco alcalino, en el lado más ácido,
suceso llamado retención de iones. Tales consideraciones tienen consecuencias obvias en la absorción y la excreción
de medicamentos, como se mostrará más específicamente en
párrafos siguientes. El surgimiento de gradientes de concentración de electrólitos débiles a través de membranas con un gradiente de pH es un proceso meramente físico y no requiere
de un sistema de transporte activo del electrolito. Todo lo que
se requiere es una membrana con permeabilidad preferencial
por una forma de un electrólito débil y un gradiente de pH
entre uno y otro lados de ella. Empero, el establecimiento del
gradiente de pH es un proceso activo.
Transporte por la membrana mediado por transportadores. La difusión
pasiva a través de la capa doble es el mecanismo predominante en la eliminación de casi todos los fármacos, aunque también pueden intervenir de
modo importante mecanismos mediados por transportadores. El transporte
activo se caracteriza por la necesidad de energía, desplazamiento contra un
gradiente electroquímico, capacidad de saturación, selectividad e inhibición competitiva por compuestos transportados conjuntamente. La trifosfatasa de adenosina (adenosine triphosphatase, ATPasa) de Na1,K1 es un
mecanismo de transporte activo. En el transporte activo secundario se utiliza la energía electroquímica almacenada en un gradiente para desplazar
a otra molécula en contra de un gradiente de concentración; por ejemplo,
la proteína de intercambio Na1-Ca21 utiliza la energía almacenada en el
gradiente de Na1 establecida por la ATPasa de Na1,K1 para exportar
Ca21 citosólico y mantenerlo a un nivel basal bajo, de aproximadamente
100 nM en la mayor parte de las células (véase cap. 33); de manera similar,
los transportadores de glucosa acoplados al Na1 SGLT1 y SGLT2 desplazan glucosa a través de la membrana del epitelio digestivo y los túbulos
renales combinando el transporte de glucosa con el fl ujo descendente de
Na1. El término difusión facilitada describe un método de transporte a
través de un portador en el que no existe aporte de energía y, por lo tanto,
el desplazamiento de la sustancia implicada se realiza a lo largo de un gradiente electroquímico como sucede en la penetración de glucosa a través
de una membrana celular de un músculo gobernada por la proteína transportadora de glucosa sensible a la insulina (glucose transporter, GLUT4).
Estos mecanismos, que en ocasiones son muy selectivos para determinada
configuración estructural de un fármaco, participan en el transporte de los
compuestos endógenos cuya velocidad de transporte por medio de difusión
pasiva sería demasiado lenta. En otros casos, funcionan como sistemas
de barrera para proteger a las células de ciertas sustancias potencialmente
tóxicas. Existen transportadores importantes desde el punto de vista farmacológico que gobiernan ya sea la entrada o la salida de los fármacos y a menudo facilitan el transporte vectorial a través de las células polarizadas. Un
transportador de salida importante que existe en diversas ubicaciones es
la glucoproteína P codificada por el gen de multirresistencia 1 (multidrug
resistance-1, MDR1). La glucoproteína P del enterocito limita la absorción
bucal de los fármacos transportados puesto que exporta compuestos de
nuevo hacia el aparato digestivo una vez que son absorbidos por difusión
pasiva. Esta misma glucoproteína puede conferir resistencia a algunas
sustancias utilizadas en la quimioterapia del cáncer (véase cap. 51). La
importancia de la glucoproteína P en la eliminación de ciertos fármacos
resalta por la presencia de polimorfi smos genéticos en MDR1 (véanse
caps. 2 y 4 y Marzolini et al., 2004) que pueden modifi car la concentración terapéutica de los fármacos. En el capítulo 2 se describen con mayor
detalle los transportadores y su participación en la acción farmacológica.
ABSORCIÓN, BIODISPONIBILIDAD Y VÍAS DE ADMINISTRACIÓN DE LOS FÁRMACOS
La absorción alude al peso de un fármaco desde el sitio de
su administración hasta el compartimiento central (fig. 1-1)

y la medida en que esto ocurre. Para las presentaciones só-
lidas, primero es necesario que la tableta o cápsula se disuelva liberando el fármaco para que se absorba. Los mé-
dicos se preocupan más por la biodisponibilidad que por la
absorción. Se llama biodisponibilidad al grado fraccionario
en que una dosis de fármaco llega a su sitio de acción, o un
líquido biológico desde el cual tiene acceso a dicho sitio. Por
ejemplo, un medicamento administrado por vía oral debe ser
absorbido en primer lugar en el estómago y los intestinos,
pero esto puede estar limitado por las características de presentación del producto, las propiedades fisicoquímicas del
medicamento o ambos factores. Como etapa siguiente, el
fármaco pasa por el hígado; en ese sitio puede ocurrir metabolismo, excreción por bilis o ambos fenómenos antes de
que el producto llegue a la circulación general. Sobre tales
bases, una fracción de la dosis administrada y absorbida será
inactivada o desviada antes de que llegue a la circulación
general y se distribuya a sus sitios de acción. Si es grande
la capacidad metabólica o excretora del hígado en relación
con el fármaco en cuestión, disminuirá sustancialmente su
biodisponibilidad (el llamado efecto de primer paso). Esta
disminución de la disponibilidad está en función del sitio
anatómico donde ocurre la absorción; otros factores anatómicos, fisiológicos y patológicos infl uyen en dicho parámetro (véase más adelante en este capítulo), y la selección de
la vía de administración debe basarse en el conocimiento
de tales situaciones.
Comparación entre la administración oral (enteral) y la parenteral. A
menudo el médico debe elegir la vía de administración de un compuesto
terapéutico, y es en tales circunstancias cuando asume interés fundamental el conocimiento de las ventajas y desventajas de las diferentes
vías que se utilicen para ese fi n. En el cuadro 1-1 se comparan las características de las principales vías utilizadas para lograr el efecto sistémico de un producto medicamentoso.
La vía oral constituye el medio más común para administrar medicamentos, dado que es la más inocua y la más cómoda y barata. Entre
sus desventajas están la incapacidad de absorción de algunos fármacos por sus características físicas (p. ej., solubilidad en agua), vómito
por irritación de la mucosa gastrointestinal, destrucción por enzimas
digestivas o pH gástrico muy ácido, irregularidades en la absorción o
aceleración de la evacuación en presencia de alimentos u otros medicamentos, y la necesidad de contar con la colaboración del paciente.
Además, en las vías gastrointestinales, los medicamentos pueden ser
metabolizados por enzimas de la flora intestinal, la mucosa o el hígado,
antes de que lleguen a la circulación general
La inyección parenteral de ciertos medicamentos ofrece una serie
de ventajas sobre la administración oral. En algunos casos, es indispensable administrar el fármaco por vía parenteral para suministrar su
forma activa, como sucede en el caso de los anticuerpos monoclonales
como infl iximab, anticuerpo contra el factor de necrosis tumoral α (tumor necrosis factor α, TNF-α) utilizado en el tratamiento de la artritis
reumatoide. La disponibilidad es por lo general más rápida, extensa y
predecible cuando el fármaco se administra por medio de una inyección. De esta manera es posible administrar la dosis efectiva con mayor
precisión. En el caso de una urgencia y cuando el paciente se encuentra inconsciente, no coopera o no puede retener nada por vía oral, el
tratamiento parenteral se convierte en una necesidad. Sin embargo, la
inyección de los fármacos tiene también algunas desventajas: es importante realizar una asepsia adecuada, en especial cuando el tratamiento
es prolongado, como sucede en la vía intravenosa o intratecal; algunas inyecciones son dolorosas; y en ocasiones es difícil que el paciente se inyecte a sí mismo cuando es necesario recurrir a esta medida.
Ingestión de fármacos. La absorción en las vías gastrointestinales es
regida por factores como el área de superfi cie para absorción, la corriente sanguínea en el sitio de absorción y el estado físico del medicamento (solución, suspensión o producto sólido), hidrosolubilidad y
concentración del fármaco en el sitio en que se absorbe. En el caso de
medicamentos que se encuentran en forma sólida, la rapidez de disolución puede ser el factor que limite su absorción, en especial si es poca
su hidrosolubilidad. Respecto de casi todos los fármacos, la absorción
en las vías gastrointestinales se hace a través de mecanismos pasivos,
razón por la cual hay mayor absorción cuando el producto en cuestión
está en la modalidad no ionizada y más lipófi la. Con base en el concepto de partición fundada en el pH, expuesto en la figura 1-2, cabría
predecir que los medicamentos que son ácidos débiles se absorberán
mejor en el estómago (pH de 1 a 2), que en duodeno y yeyuno (pH de
3 a 6) y la situación contraria priva en el caso de bases débiles (alcalinos). Sin embargo, el epitelio del estómago está revestido por una capa
mucosa gruesa y su área de superfi cie es pequeña; a diferencia de ello,
las vellosidades de duodeno y yeyuno poseen una enorme área superficial (aproximadamente 200 m2). Por esa razón, la rapidez e índice de
absorción de un fármaco en el intestino será mayor que en el estómago,
incluso si el medicamento se halla predominantemente ionizado en el
intestino y no lo está en el estómago (en su mayor parte). Así pues, es
probable que cualquier factor que acelere el vaciamiento del estómago,
apresurará la absorción de medicamentos, en tanto que cualquier factor que retrase el vaciamiento tiende a ejercer el efecto contrario, sean
cuales sean las características del fármaco. En la mujer, los estrógenos
actúan sobre el vaciamiento gástrico (es decir, es más lento en las mujeres premenopáusicas y en las mujeres que toman estrógenos como
tratamiento sustitutivo que en los varones).
Los medicamentos que son destruidos por las secreciones gástricas
o que provocan irritación gástrica se administran con una capa entérica
que impide su disolución en el contenido gástrico ácido. No obstante,
algunos preparados con capa entérica también son resistentes a la disolución intestinal, lo que reduce su absorción. Sin embargo, la capa
entérica es de gran utilidad para algunos fármacos como la aspirina, que
en muchos pacientes provoca irritación gástrica importante.
Preparados de liberación controlada. La velocidad de absorción de
un producto medicinal que se administra en forma de tableta o en otra
presentación sólida para ingestión, depende en parte de su velocidad
de disolución en los líquidos gastrointestinales. El factor mencionado
constituye la base para preparar los fármacos llamados de liberación
controlada, extendida, sostenida o de acción prolongada, que puedan
absorberse de modo lento y uniforme durante 8 h o más. Existen preparados de este tipo en todas las categorías principales de fármacos. Sus
ventajas potenciales son la menor frecuencia de administración que las
presentaciones convencionales (lo que quizá mejora la aceptación por
parte del paciente), el efecto terapéutico constante durante la noche y
la menor frecuencia o intensidad de efectos indeseables (al eliminar los
picos en la concentración del fármaco) y de concentración sanguínea
no terapéutica (al eliminar las concentraciones mínimas) que a menudo
ocurren al administrar una presentación de liberación inmediata.
Muchos preparados de liberación lenta cumplen con estas expectativas y se prefi eren en ciertas situaciones terapéuticas, como el tratamiento antidepresivo (Nemeroff, 2003) o el tratamiento con el antagonista
del calcio dihidropiridina (véase cap. 32). Sin embargo, estos productos
tienen algunos inconvenientes. En general, las variaciones entre los diversos pacientes en términos de la concentración que alcanza el fármaco
son mayores cuando se utilizan presentaciones de liberación controlada que con las de liberación inmediata. Al repetir la administración,
la concentración mínima del fármaco con una presentación de liberación
controlada no siempre difi ere de la que se observa con los preparados
de liberación inmediata, si bien el intervalo entre las concentraciones
mínimas es mayor con los productos bien diseñados de liberación lenta.
La forma farmacéutica en ocasiones falla y origina una “descarga masiva de la dosis” con los efectos secundarios resultantes, ya que la dosis total del fármaco que se ingiere en determinado momento es varias
veces mayor que la cantidad contenida en la preparación convencional.
Algunos de los factores que contribuyen a la “liberación inmediata” de
ciertos preparados de liberación controlada son la acidez gástrica y el
hecho de administrarlos con un alimento grasoso. Las presentaciones
de liberación controlada son ideales para los medicamentos con una
semivida corta (,4 h). Sin embargo, también existen preparados llamados de liberación controlada para medicamentos con una semivida más
prolongada (.12 h). Estos productos son casi siempre más caros y no se
deben prescribir a menos que se hayan demostrado sus ventajas.
Administración sublingual. La absorción a partir de la mucosa oral es
importante para algunos medicamentos a pesar de que la superficie
es pequeña. El drenaje venoso de la boca se dirige hacia la vena cava
superior, lo que protege al fármaco de un metabolismo hepático rápido
de primer paso. Por ejemplo, la nitroglicerina es efectiva en su presentación sublingual puesto que no es iónica y es altamente liposoluble.
Por lo tanto, se absorbe con gran rapidez. Además, es muy potente; se
necesita una cantidad relativamente pequeña de moléculas para producir el efecto terapéutico. Si una tableta de nitroglicerina se deglute, el
metabolismo hepático basta para impedir su aparición en la circulación
general.
Absorción transdérmica. No todos los fármacos penetran con facilidad
a través de la piel íntegra. La absorción de los que penetran depende de
la superfi cie sobre la que se aplican y de su liposolubilidad, ya que la
epidermis se comporta como lipobarrera (véase cap. 63). Sin embargo,
la dermis es permeable a numerosos solutos y, por lo tanto, los fármacos
se absorben con mayor facilidad hacia la circulación general a través de
la piel desnuda, quemada o lacerada. Asimismo, la infl amación y otras
circunstancias que aumentan la irrigación cutánea también incrementan la absorción. Cuando una sustancia altamente liposoluble (p. ej.,
un insecticida liposoluble en un solvente orgánico) se absorbe a través
de la piel algunas veces produce efectos tóxicos. Es posible aumentar
la absorción a través de la piel suspendiendo el fármaco en un vehículo oleoso y frotando la preparación sobre la piel. La piel hidratada es
más permeable que la piel seca, de manera que conviene modificar la
presentación o aplicar un vendaje oclusivo para facilitar la absorción.
Cada vez son más populares los parches de liberación controlada, como
el de nicotina para dejar de fumar, la escopolamina para la cinetosis, la
nitroglicerina para la angina de pecho, la testosterona y los estróge-nos
para la terapéutica sustitutiva y diversos estrógenos y progestágenos
para el control prenatal.
Administración rectal. La vía rectal suele ser útil cuando la ingestión
del medicamento resulta imposible a causa de vómito o inconsciencia del enfermo, una situación relevante en particular para niños peque-
ños. Cerca de 50% del fármaco que se absorbe por el recto “salvaría
la barrera hepática”; de este modo, la posibilidad de metabolismo de
primer paso por dicho órgano es menor que con una dosis ingerida. Sin
embargo, la absorción por el recto suele ser irregular e incompleta, y
muchos fármacos irritan la mucosa de dicho órgano.
Inyección parenteral. Las formas principales de aplicación parenteral
son intravenosa, subcutánea e intramuscular. En el caso de las vías subcutánea e intramuscular, la absorción ocurre por difusión sencilla, al seguir
el gradiente que media entre el depósito del medicamento y el plasma.
La velocidad depende del área de las membranas capilares que absorben
el producto y de la solubilidad de la sustancia en el líquido intersticial.
Los canales acuosos relativamente grandes de la membrana endotelial
permiten una difusión indiscriminada de moléculas, independiente de su
liposolubilidad. Las moléculas grandes, como las de las proteínas, penetran con lentitud en la circulación a través de los conductos linfáticos.
Los fármacos que se administran por cualquier vía (excepto la intraarterial) en la circulación general, están sujetos a una posible eliminación de primer paso por los pulmones, antes de distribuirse al resto
del cuerpo. Los pulmones son sitio temporal de eliminación de diversos
medicamentos, en particular los que son bases débiles y están predominantemente no ionizados en el pH de la sangre, al parecer por su partición en lípidos. Los pulmones también sirven como fi ltro de partículas
que pueden introducirse por vía intravenosa y, por supuesto, son un
medio para la eliminación de sustancias volátiles.
Vía intravenosa. La inyección intravenosa de fármacos en solución
acuosa evita los factores relevantes que intervienen en la absorción,
porque en la sangre venosa la biodisponibilidad es completa y rápida.
Asimismo, la llegada del producto a los tejidos se hace de manera controlada y con una exactitud y celeridad que no son posibles por otras
vías. En algunos casos, como en la inducción de anestesia quirúrgica, la
dosis del fármaco no se determina de antemano, sino que se ajusta a las
reacciones del enfermo. De la misma manera, sólo por vía intravenosa
pueden administrarse algunas soluciones irritantes, porque el fármaco,
si se inyecta despacio, se diluye en gran medida en la sangre.
Esta vía de administración ofrece tanto ventajas como desventajas.
En primer lugar, puede haber reacciones adversas cuando la concentración alta del fármaco llega con rapidez al plasma y los tejidos. Sin
embargo, existen varias circunstancias terapéuticas en las que conviene
administrar el fármaco por medio de un bolo (un pequeño volumen que
se administra rápidamente, p. ej., el activador del plasminógeno hístico [tisular] inmediatamente después de un infarto agudo del miocardio) y otros casos en los que el fármaco se debe administrar con mayor
lentitud, como los que se aplican diluidos por vía intravenosa (p. ej.,
antibióticos). Cuando se administra un fármaco por vía intravenosa es
importante vigilar la respuesta del paciente. Además, una vez que se inyecta el medicamento, no hay marcha atrás. La posibilidad de repetir las
inyecciones intravenosas depende de la posibilidad de mantener la vena
permeable. Los fármacos en un vehículo aceitoso, los que precipitan los
componentes sanguíneos o hemolizan a los eritrocitos y las combinaciones de medicamentos que provocan la formación de precipitados no se
deben administrar por esta vía.
Subcutánea. Un fármaco se inyecta por vía subcutánea sólo cuando
no irrita los tejidos; de lo contrario provoca dolor intenso, necrosis y
desprendimiento de los tejidos. Después de una inyección subcutánea,
la velocidad de absorción del fármaco suele ser lo suficientemente constante y lenta como para proporcionar un efecto sostenido. Además, el
periodo de absorción se puede modifi car intencionalmente, como sucede con la insulina, según el tamaño de las partículas, los complejos proteínicos y el pH para obtener una preparación de acción corta (3 a 6 h),
intermedia (10 a 18 h) o prolongada (18 a 24 h). La absorción también
se prolonga si se incorpora un vasoconstrictor a la solución del medicamento que se inyecta por vía subcutánea. De esta manera, el anestésico
local inyectable lidocaína contiene adrenalina en su presentación. La
absorción de los fármacos que se depositan bajo la piel dentro de una
microesfera es lenta y se prolonga varias semanas o meses; algunas hormonas (p. ej., anticonceptivos) se administran de esta manera.
Intramuscular. Los fármacos en solución acuosa se absorben con rapidez después de su inyección intramuscular, pero la velocidad depende
de la circulación en el sitio de la inyección. Esta velocidad se puede
regular hasta cierto grado por medio de calor local, masaje o ejercicio.
Por ejemplo, la absorción de la insulina suele ser más rápida cuando se
inyecta en el brazo y la pared abdominal que en el muslo; sin embargo,
el hecho de correr o trotar provoca en ocasiones el descenso repentino
de la glucemia cuando la insulina se inyecta en el muslo en lugar de
utilizar el brazo o la pared abdominal, ya que este ejercicio aumenta la
circulación de la pierna. El baño caliente acelera la absorción en todos
estos sitios a causa de la vasodilatación. En general, la velocidad con la
que un fármaco se absorbe después de su inyección en una preparación
acuosa aplicada en el deltoides y vasto lateral es más rápida que cuando
se aplica en el glúteo mayor. En la mujer, la velocidad es especialmente lenta cuando la inyección se aplica en el glúteo mayor. Al parecer,
la razón es la distribución distinta de la grasa subcutánea en el varón
y la mujer, además de que la grasa posee una circulación deficiente.
Algunos pacientes muy obesos o emaciados exhiben patrones raros de
absorción después de la inyección intramuscular o subcutánea. Para que
la absorción de un medicamento intramuscular sea lenta y constante se
debe aplicar una solución en aceite o suspendida en algún otro vehículo
de depósito. Los antibióticos suelen administrarse de esta forma. Las
sustancias que son demasiado irritantes como para administrarlas por
vía subcutánea algunas veces pueden aplicarse por vía intramuscular.
Vía intraarterial. En ocasiones se inyecta directamente un medicamento en una arteria para limitar su efecto a un tejido u órgano particular, por
ejemplo en el tratamiento de tumores hepáticos y de cabeza y cuello. A
veces se administran por esta vía ajustes diagnósticos (p. ej., albúmina sé-
rica humana marcada con tecnecio). La inyección dentro de una arteria
requiere enorme cuidado y debe ser del dominio de expertos. Cuando los
medicamentos se aplican por vía intraarterial, se pierde el metabolismo de
primer paso y los efectos depuradores de los pulmones.
Vía intrarraquídea. La barrera hematoencefálica y la que separa
sangre y líquido cefalorraquídeo (LCR) impiden o retrasan la penetración de fármacos al sistema nervioso central (SNC). Por tanto, si se
pretende obtener efectos locales y rápidos en las meninges o el eje cefalorraquídeo (cerebroespinal), como ocurre en la raquianestesia o en
infecciones agudas del sistema nervioso central, a veces se inyectan los
fármacos de manera directa en el espacio subaracnoideo raquídeo. Los
tumores encefálicos también pueden tratarse por medio de administración intraventricular directa de medicamentos.
Absorción en pulmones. A condición de que no originen irritación, fármacos gaseosos y volátiles pueden inhalarse y absorberse en el epitelio pulmonar y las mucosas de las vías respiratorias. Por este medio, el producto
llega pronto a la circulación, dado que el área de superficie pulmonar es
grande. En los capítulos 13 y 15 se enuncian los principios que rigen la
absorción y la excreción de anestésicos y otros gases terapéuticos.
Además, es posible pulverizar las soluciones de medicamentos y así
inhalar las fi nísimas gotitas (aerosol). Entre las ventajas de esta forma de
administración destacan la absorción casi instantánea del fármaco en sangre, la eliminación de las pérdidas de primer paso por el hígado y, en el
caso de neumopatías, la aplicación local del producto en el sitio de acción
buscado. Por ejemplo, de esta manera pueden suministrarse medicamentos para el tratamiento de la rinitis alérgica o el asma bronquial (véase
cap. 27). La absorción por pulmones constituye también un mecanismo
importante de penetración de algunas drogas ilícitas y tóxicos ambientales de composición y estado físico diversos. Después de la inhalación,
surgen a veces reacciones locales y sistémicas a sustancias alergenas.
Aplicación local (tópica). Mucosas. Se aplican fármacos también en las
mucosas de conjuntiva, nasofaringe, bucofaringe, vagina, colon, uretra
y vejiga, con el fi n de lograr efectos locales. En ocasiones, como ocurre
con la aplicación de la hormona antidiurética sintética en la mucosa
nasal, se busca ante todo la absorción generalizada. La absorción por
mucosas se produce con gran rapidez. De hecho, los anestésicos locales
que se utilizan para obtener algún efecto en el propio sitio de aplicación a veces se absorben con tal rapidez que ejercen efectos tóxicos
generalizados.
Ojo. Los fármacos oftálmicos de uso local se utilizan por sus efectos en el sitio de aplicación (véase cap. 63). Por lo general, es indeseable
la absorción sistémica que resulta del drenaje por el conducto nasolagrimal. Los fármacos que se absorben a través del drenaje ocular no son
metabolizados en el hígado, de manera que la administración oftálmica
de gotas de antiadrenérgicos β o corticosteroides puede originar efectos
indeseables. Para que se produzcan efectos locales es necesario que el
fármaco se absorba a través de la córnea; por lo tanto, las infecciones
o traumatismos corneales aceleran la absorción. Los sistemas que prolongan la duración de la acción (p. ej., suspensiones y pomadas) son
de gran utilidad en el tratamiento oftálmico. Los implantes oculares,
como las inclusiones con pilocarpina para el tratamiento del glaucoma,
ofrecen la aplicación continua de una pequeña cantidad del fármaco. Se
pierde muy poco a través del drenaje ocular y, por lo tanto, sus efectos
colaterales sistémicos se reducen al mínimo.
Nuevos métodos de liberación de drogas. Se están utilizando endopró-
tesis y otros dispositivos eluyentes de fármacos para aplicar el medicamento en forma circunscrita y reducir al mínimo su contacto con la
circulación general. Los efectos adversos de varios compuestos importantes se reducen considerablemente si se combinan con una serie de
vehículos que modifi can su distribución. Por ejemplo, el citotóxico caliqueamicina, al combinarse con un anticuerpo contra el antígeno ubicado
en la superfi cie de ciertas células leucémicas, dirige al fármaco hacia su
sitio de acción, mejorando el índice terapéutico de la caliqueamicina.
Los avances más recientes en el campo de las vías de administración
comprenden el empleo de polímeros biocompatibles fi jados a monó-
meros funcionales adheridos de manera tal que permiten la unión de
moléculas del fármaco hasta el polímero.
Para elaborar un conjugado de un polímero que se convierta en un
profármaco estable y de circulación prolongada se altera el peso molecular del polímero y el clivaje entre el fármaco y el polímero. Este
enlace se creó para mantener al fármaco inactivo hasta que es liberado del polímero por algún factor desencadenante específi co para cada
enfermedad, por lo general la actividad enzimática sobre el tejido que
suministra el fármaco activo en el sitio de la patología.
Bioequivalencia. Los productos medicamentosos se consideran como
equivalentes farmacéuticos si contienen los mismos ingredientes activos y tienen potencia o concentración, presentación y vías de administración idénticas. Dos sustancias farmacéuticamente equivalentes se
consideran bioequivalentes si la rapidez y magnitud de la biodisponibilidad del ingrediente activo en ambos no difiere en mayor grado en las
situaciones idóneas de “prueba”. En el pasado, a veces se detectaban
diferencias en la biodisponibilidad de las presentaciones elaboradas por
fabricantes distintos, e incluso en lotes diferentes de productos de un
solo fabricante. Estas diferencias se observaban sobre todo en las presentaciones orales de ciertos fármacos poco solubles y de absorción
lenta, como el antibiótico urinario metronidazol (FLAGYL). Al principio,
la forma genérica no era bioequivalente, puesto que el fabricante no
podía simular el proceso original utilizado para comprimir el fármaco
con el fin de facilitar su absorción. Algunas diferencias en la forma de
los cristales, el tamaño de las partículas y otras características típicas
del fármaco que no se regulan de manera rigurosa durante la formulación y elaboración alteran la desintegración de la forma farmacéutica
y la disolución del fármaco, lo que modifica la velocidad y el grado de
absorción.
La posible falta de equivalencia de diversos preparados medicamentosos
ha sido motivo de preocupación. Gracias a exigencias normativas cada vez
más enérgicas, hay pocos casos corroborados (y quizá ninguno) de falta de
equivalencia entre productos medicamentosos aprobados en años recientes.
La importancia de una posible falta de equivalencia entre fármacos se explica en mayor detalle en relación con la nomenclatura de los medicamentos y
la elección de un nombre en la elaboración de recetas (véase Apéndice I).
DISTRIBUCIÓN DE LOS FÁRMACOS
Después de su absorción o administración en el torrente circulatorio general, un fármaco se distribuye en los líquidos intersticial e intracelular. Tal fenómeno expresa muy diversos
factores fi siológicos y las propiedades fisicoquímicas particulares de cada producto medicamentoso. Elementos que
rigen la rapidez de “llegada” y la posible cantidad del fármaco que se distribuye en los tejidos son el gasto cardíaco,
la corriente sanguínea regional y el volumen hístico. En el
comienzo, hígado, riñones, encéfalo y otros órganos con gran
riego sanguíneo reciben la mayor parte del medicamento, en
tanto que es mucho más lenta la llegada del mismo a músculos, casi todas las vísceras, piel y grasa. Esta fase de “segunda distribución” quizá necesite minutos a horas para que la
concentración del fármaco en los tejidos entre en una fase
de equilibrio por distribución, con la que hay en sangre. La
segunda fase también incluye una fracción mucho mayor de
la masa corporal, que la fase inicial, y por lo común explica
gran parte de la distribución extravascular del medicamento.
Con excepciones como el encéfalo, la difusión del fármaco
en el líquido intersticial se hace con gran rapidez, por la naturaleza altamente permeable de la membrana del endotelio
capilar. Por tal razón, la distribución en tejidos depende de la
partición del fármaco entre la sangre y el tejido particular. La
liposolubilidad es el factor determinante de dicha captación,
como también lo es cualquier gradiente de pH entre los líquidos intracelular y extracelular en el caso de medicamentos
que son ácidos o bases débiles. Sin embargo, en general, no
es grande la retención de iones que se vincula con este último
factor, dado que la diferencia de pH (7.0 en comparación con
7.4) es pequeña. El factor determinante de mayor cuantía en
la partición sangre/tejido es la unión relativa del medicamento a las proteínas plasmáticas y macromoléculas tisulares.
Proteínas plasmáticas. Muchos fármacos circulan en el
torrente sanguíneo unidos a ciertas proteínas plasmáticas. La
albúmina es un transportador fundamental para los fármacos
ácidos; la glucoproteína ácida α1 se une a ciertos fármacos bá-
sicos. El enlace inespecífi co con otras proteínas plasmáticas
es mucho menos común. Esta unión es casi siempre reversible; en algunos casos se produce una unión covalente de ciertos medicamentos reactivos, como las sustancias alquilantes.
Además de unirse con una serie de proteínas transportadoras
como albúmina, ciertos fármacos se enlazan con proteínas
que funcionan como transportadoras de ciertas hormonas,
como el enlace de los estrógenos o la testosterona con la
globulina fi jadora de hormonas sexuales o el enlace de
la hormona tiroidea con la globulina fi jadora de tiroxina.
Del total del fármaco, la fracción plasmática que habrá de
unirse dependerá de la concentración de aquél, su afinidad
por los sitios de unión y el número de estos últimos. Son las
relaciones entre acción y masa lo que determina las concentraciones de producto unido y no unido (véase más adelante
en este capítulo). Si la concentración es pequeña (menor que
la constante de disociación de unión a proteínas plasmáticas), la fracción ligada estará en función del número de sitios de unión y de la constante de disociación. En caso de
haber grandes concentraciones del fármaco (que excedan de
la constante de disociación), la fracción ligada estará en función del número de sitios de unión y de la concentración del
medicamento. Por consiguiente, la unión a proteínas plasmá-
ticas es un fenómeno saturable y no lineal. Sin embargo, para
algunos medicamentos son limitados los márgenes terapéuticos de las concentraciones plasmáticas; por consiguiente, la
magnitud de la unión del fármaco y la fracción libre o no ligada son relativamente constantes. Los porcentajes incluidos
en el Apéndice II incluyen sólo la situación anterior, salvo
que se indique lo contrario. La extensión de la unión a proteínas plasmáticas también puede ser modifi cada por factores
propios de enfermedades. Por ejemplo, la hipoalbuminemia
que es consecuencia de una hepatopatía grave o de síndrome
nefrótico disminuye la unión, y aumenta así la fracción libre.
Asimismo, los trastornos que originan una reacción de fase
aguda (cáncer, artritis, infarto de miocardio, enfermedad de
Crohn) permiten que se incrementen los valores de glucoproteína ácida α1 y se genere una mayor unión de fármacos
alcalinos (básicos).
La unión entre fármacos y proteínas plasmáticas del tipo de la albúmina no es selectiva, y el número de sitios de unión es relativamente
grande (gran capacidad), de manera que muchos medicamentos con
características fisicoquímicas similares pueden competir entre ellos y
con varias sustancias endógenas por estos sitios de enlace, lo que tiene como resultado el desplazamiento notable de un fármaco por otro.
Por ejemplo, las sulfonamidas y otros aniones orgánicos desplazan a la
bilirrubina no conjugada de su sitio de enlace con la albúmina, lo que
aumenta el riesgo de encefalopatía por bilirrubina en el recién nacido.
Sin embargo, los efectos indeseables por competencia entre fármacos
por los sitios de enlace no constituyen una preocupación clínica importante en la mayor parte de los casos. Las respuestas a los medicamentos, tanto efectivas como adversas, son función de la concentración del
fármaco libre, de manera que la concentración en estado estacionario
del medicamento libre cambia de manera notable sólo cuando cambia
el ingreso del fármaco (velocidad de administración) o cambia la depuración de la droga no unida (véanse la ecuación 1-2 y la descripción
más adelante). Por lo tanto, la concentración en estado estacionario del
fármaco libre es independiente del grado de unión con las proteínas. No
obstante, para los fármacos con un índice terapéutico reducido, algunas
veces es preocupante un cambio en la concentración de medicamento
libre inmediatamente después de administrar un medicamento que genere desplazamiento, como sucede con el anticoagulante warfarina. Un
problema más común originado por la competencia de fármacos por los
sitios de unión en las proteínas plasmáticas es la interpretación errónea
de la concentración plasmática de un fármaco, ya que la mayor parte de
los análisis no distingue al medicamento libre del adherido.
Es importante señalar que la unión de un fármaco con la proteína
plasmática reduce su concentración en los tejidos y en el sitio de acción, puesto que únicamente el fármaco libre se encuentra en equilibrio
a través de las membranas. Por consiguiente, una vez que se logra una
distribución equilibrada, la concentración del fármaco libre y activo
en el agua intracelular es igual a la del plasma, con excepción de los
casos en que existe transporte por medio de transportadores. La unión
entre fármacos y proteínas plasmáticas también limita la filtración
glomerular del medicamento, puesto que este proceso no cambia de
inmediato la concentración de fármaco libre en el plasma (también se
filtra agua). Sin embargo, la unión con las proteínas plasmáticas no
limita la secreción tubular renal ni la biotransformación, ya que estos
procesos reducen la concentración de fármaco libre, e inmediatamente
después el medicamento se separa del complejo fármaco-proteína, con
lo que se restablece el equilibrio entre el fármaco libre y el adherido.
La unión con las proteínas plasmáticas también reduce el transporte y el
metabolismo del fármaco, con excepción de los casos en que éstas son
especialmente eficientes, y la eliminación del fármaco, que se calcula
con base en el fármaco libre, excede la circulación plasmática en los
órganos.
Fijación hística (tisular). Muchos medicamentos se acumulan en los tejidos en concentraciones mayores que en líquidos
extracelulares y sangre. Por ejemplo, durante la administración duradera del antipalúdico quinacrina, la concentración
de este fármaco en el hígado puede ser cientos de veces mayor que la que hay en sangre. Dicha acumulación tal vez sea
consecuencia del transporte activo, o comúnmente, de la
unión. La unión a tejidos por lo regular sucede con componentes celulares como proteínas, fosfolípidos o proteínas nucleares, y casi siempre es reversible. Una fracción importante
del fármaco dentro del organismo puede fi jarse y quedar ligada a esta forma, y constituir un “reservorio” que prolongue
la acción del medicamento en el mismo tejido o en un sitio distante a través de la circulación. Este tipo de fijación y
acumulación en los tejidos también origina efectos adversos
locales, por ejemplo, cuando se acumula el aminoglucósido
gentamicina en el riñón y el sistema vestibular.
La grasa como depósito. Muchos fármacos liposolubles se almacenan por solución en la grasa neutra. En personas obesas puede llegar a
50% el contenido de lípidos del cuerpo, e incluso en casos de inanición
sigue siendo 10% del peso corporal; por tanto, la grasa constituye un
depósito importante de productos liposolubles. Por ejemplo, hasta 70%
del tiopental, barbitúrico fuertemente liposoluble, puede hallarse en la
grasa corporal 3 h después de administrado. La grasa es un reservorio
bastante estable gracias a su irrigación tan reducida. Sin embargo, entre
los fármacos altamente lipófi los (p. ej., remifentanilo y algunos bloqueadores β) el grado de lipofilia no pronostica su distribución en los
obesos.
Hueso. Las tetraciclinas (como otros compuestos quelantes de iones metálicos divalentes) y los metales pesados se acumulan en el hueso
por adsorción en la superfi cie cristalina de dicho tejido e incorporación
final a la trama cristalina. El hueso puede convertirse en un depósito de
liberación lenta de agentes tóxicos, como el plomo o el radio, a la sangre; tales efectos tal vez persistan mucho después de que cesó la exposición o contacto. La destrucción local de la médula ósea también reduce
la irrigación y prolonga el efecto de depósito, porque el agente tóxico
queda separado e independiente de la circulación, lo cual puede agravar más el daño local directo al hueso. De este modo, se establece un
círculo vicioso en el que, cuanto mayor sea la exposición al agente
tóxico, tanto más lenta será su eliminación. La adsorción del fármaco
en la superfi cie cristalina del hueso y su incorporación en la trama cristalina ofrece una serie de ventajas terapéuticas en el tratamiento de la
osteoporosis. Los fosfonatos como el etidronato de sodio se fijan con
fuerza a los cristales de hidroxiapatita en la matriz ósea mineralizada.
Sin embargo, a diferencia de los pirofosfonatos naturales, el etidronato
es resistente a la degradación que originan las pirofosfatasas y, por lo
tanto, estabiliza a la matriz ósea.
Redistribución. Por lo regular, la terminación del efecto de
un fármaco ocurre por biotransformación y excreción, pero eso
también puede ser consecuencia de la redistribución de aquél
desde el sitio de acción hacia otros tejidos o lugares. Cuando
un producto fuertemente liposoluble, con acción en el encéfalo o el aparato cardiovascular, se administra de forma rápida
mediante inyección intravenosa o por inhalación, la redistribución es el factor que más contribuye a la terminación del efecto medicamentoso. Un buen ejemplo de lo anterior sería el uso
del tiopental, un anestésico intravenoso que es fuertemente
liposoluble. La corriente sanguínea al encéfalo es muy grande,
razón por la cual el fármaco llega a su concentración máxima
en dicho órgano en término de 1 min de haber sido inyectado en
la vena. Una vez terminada la inyección, la concentración plasmática disminuye porque el tiopental se difunde a otros tejidos
como el músculo. La concentración del medicamento en el encéfalo “corresponde” a la del plasma debido a la escasa unión
del medicamento a los componentes encefálicos. Por tal razón,
la anestesia comienza a muy breve plazo y también concluye
de ese mismo modo. Ambos hechos guardan relación directa
con la concentración del fármaco en el encéfalo.
Sistema nervioso central (SNC) y líquido cefalorraquí-
deo (LCR). La distribución de fármacos en el sistema nervioso central, a partir de la sangre, es un fenómeno peculiar.
Una razón de lo anterior es que las células del endotelio de
capilares encefálicos muestran uniones estrechas continuas
y, como consecuencia, la penetración de fármacos al tejido
encefálico depende del transporte transcelular y no del paracelular. Las características peculiares de las células gliales
pericapilares también contribuyen a la barrera hematoencefálica. En el plexo coroideo, de la misma manera existe una
barrera de sangre y líquido cefalorraquídeo (LCR) semejante, excepto en que son las células epiteliales las que están
juntas por medio de uniones estrechas, y no las células del
endotelio. Como consecuencia, la liposolubilidad de las formas no ionizada y libre del fármaco constituyen un factor
determinante de su captación por el encéfalo, es decir, cuanto más lipófi lo sea, con mayor facilidad cruzará la barrera
hematoencefálica. Esta situación se utiliza a menudo en el
diseño de fármacos para modifi car su distribución encefálica; por ejemplo, los llamados antihistamínicos de segunda
generación como loratidina alcanzan una concentración encefálica mucho menor que los medicamentos del tipo de la
difenhidramina y, por lo tanto, no son sedantes. Algunos
fármacos penetran en el SNC gracias a ciertos transportadores de la captación que normalmente participan en el transporte de nutrientes y compuestos endógenos desde la sangre
hasta el cerebro y el líquido cefalorraquídeo.
Otro factor importante en la barrera hematoencefálica
son los transportadores de membrana, que acarrean material
que sale y se encuentran en las células endoteliales capilares
encefálicas; expulsan de la célula a un gran número de fármacos distintos desde el punto de vista químico. Dos de las
más importantes son la glucoproteína P ([P-glycoprotein, Pgp], codifi cada por el gen MDRI) y el polipéptido transportador de aniones orgánicos (organic anion-transporting polypeptide, OATP). Actúan restringiendo de manera notable
el acceso del fármaco al tejido que expresa el transportador
de efl ujo. Juntos, P-gp y la familia de los OATP expulsan a
una gran variedad de fármacos con estructura distinta (Kim,
2003) (véase cap. 2). La expresión de isoformas de OATP en
el aparato digestivo, hígado, riñón y la barrera hematoencefálica tiene gran importancia para la absorción y eliminación
de fármacos, así como para su penetración en los tejidos. La
expresión de estos transportadores de efl ujo explica el acceso relativamente restringido de los fármacos en el cerebro y
otros tejidos, como los testículos, donde la concentración farmacológica es menor que la necesaria para lograr un efecto
deseado a pesar de que la irrigación es adecuada. Esta situación ocurre con los inhibidores de la proteasa de VIH y con
la loperamida, opiáceo potente con actividad sistémica que
carece de los efectos centrales característicos de otros opiáceos
(véase cap. 21). También existen transportadores de eflujo
que secretan de manera activa al fármaco del líquido cefaloraquídeo hacia la sangre en el plexo coroideo (véanse los caps.
2 y 3 para mayores detalles sobre la contribución de los transportadores de fármacos a la función de barrera). Otras veces
los fármacos abandonan el sistema nervioso central mediante
el importante fl ujo de líquido cefalorraquídeo a través de las
vellosidades aracnoideas. En general, la función de la barrera
hematoencefálica se conserva bastante bien, pero la inflamación meníngea y encefálica aumentan la permeabilidad local.
En fecha reciente se introdujo la desorganización de la barrera
hematoencefálica como tratamiento de ciertos tumores cerebrales, como linfoma primario del sistema nervioso central (Tyson et al., 2003). La finalidad de este tratamiento es mejorar la
distribución de la quimioterapia en el tumor cerebral al tiempo
que se conserva la función cognoscitiva, que suele dañarse con
la radioterapia convencional (Dahlborg et al., 1998).
Transferencia placentaria de fármacos. La transferencia de fármacos a través de la placenta tiene especial importancia puesto que muchos medicamentos causan anomalías
en el feto. Si los medicamentos se administran inmediatamente antes del parto, como suele suceder con los tocolíticos
en el tratamiento del trabajo de parto de pretérmino, también
tienen efectos adversos sobre el neonato. Algunos de los factores generales que afectan la transferencia de los fármacos
a través de la placenta son su liposolubilidad, su grado de
fijación plasmática y el grado de ionización de los ácidos y
bases débiles. El plasma fetal es ligeramente más ácido que
el materno (pH de 7.0 a 7.2 contra 7.4), de manera que los
fármacos alcalinos sufren atrapamiento iónico crónico. Al
igual que en el cerebro, existen P-gp y otros transportadores
en la placenta, y funcionan restringiendo el contacto entre el
feto y las sustancias potencialmente tóxicas. Por otro lado, la creencia de que la placenta constituye una barrera absoluta
para los fármacos es completamente errónea (Holcberg et al.,
2003), en parte puesto que también existen varios transportadores que median el infl ujo de drogas (Unadkat et al., 2004).
Hasta cierto grado, el feto se encuentra expuesto a todos los
fármacos que consume la madre.
EXCRECIÓN DE FÁRMACOS
Los fármacos se eliminan del organismo sin cambios, mediante el proceso de excreción, o se transforman en metabolitos. Los órganos excretores (después de excluir al pulmón)
eliminan compuestos polares con mayor efi cacia que sustancias de gran liposolubilidad. Por tal razón, los medicamentos
liposolubles no se eliminan de manera fácil, hasta que se metabolizan a compuestos más polares.
Los riñones son los órganos más importantes para excretar fármacos y sus metabolitos. Las sustancias eliminadas en
las heces son predominantemente medicamentos ingeridos
no absorbidos o metabolitos excretados en la bilis o secretados directamente en las vías intestinales y, por ende, que no
se resorben. La excreción de fármacos en la leche materna es
importante, no por las cantidades que se eliminan por dicho
líquido, sino porque son causa posible de efectos farmacológicos no buscados en el lactante amamantado. La excreción por
los pulmones es importante más bien para la eliminación de
gases anestésicos (véase cap. 13).
Excreción por riñones. La excreción de medicamentos y
metabolitos en la orina incluye tres procesos concretos: filtración glomerular, secreción tubular activa y resorción tubular pasiva. En general, los cambios en la función global de
los riñones modifi can los tres fenómenos en grado semejante.
Incluso en las personas sanas, la función renal es variable.
En los neonatos, la función renal es reducida si se le compara con el volumen corporal, pero madura con rapidez en los
primeros meses de vida. Durante la madurez, la función renal desciende lentamente, a una velocidad aproximada de 1%
anual, de manera que en algunos ancianos existe un deterioro
funcional importante.
La cantidad de fármaco que penetra en los túbulos por
filtración depende de la fi ltración glomerular y la magnitud
de la unión del medicamento a proteínas plasmáticas; se
filtra solamente el producto libre, es decir, no fi jado. En el
túbulo renal proximal, la secreción tubular activa mediada
por portador también puede “aportar” fármaco al líquido
tubular. Los transportadores como la glucoproteína P y la
proteína de tipo 2 vinculada con resistencia a múltiples
medicamentos (multidrug-resistance-associated protein
type 2, MRP2) localizada en el borde en cepillo apical, son
los que predominantemente hacen factible la secreción de
aniones anfi páticos y metabolitos conjugados (como glucurónidos, sulfatos y aductos de glutatión), respectivamente
(véanse caps. 2 y 3). Los sistemas de transporte del casete
enlazador a trifosfato de adenosina (ATP) (ATP-binding
cassette, ABC) que son más selectivos para fármacos canó-
nicos orgánicos intervienen en la secreción de los álcalis orgánicos (bases). Los transportadores de membrana situados
en la porción distal del túbulo renal, se encargan de la resorción activa del medicamento desde el interior del túbulo
para devolverlo a la circulación general. Sin embargo, gran
parte de la resorción en cuestión se realiza por difusión no
iónica.
En los túbulos proximales y distales, las formas no ionizadas de ácidos y bases débiles experimentan resorción
pasiva neta. El gradiente de concentración para la difusión
retrógrada es creado por la resorción de agua, con sodio
y otros iones inorgánicos. Las células de los túbulos son
menos permeables a las formas ionizadas de electrólitos
débiles, razón por la cual la resorción pasiva de tales sustancias se realiza con base en el pH (“dependiente de pH”).
Si se hace más alcalina la orina tubular, se excretan los ácidos débiles con rapidez y magnitud mayores, porque están
más ionizados y disminuye la resorción pasiva. Cuando la
orina tubular se acidifi ca, la fracción de fármaco ionizado disminuye y su excreción se reduce. La alcalinización
y acidifi cación de la orina tienen efectos opuestos sobre la
excreción de las bases débiles. En el tratamiento de la intoxicación medicamentosa, es posible acelerar la excreción
de ciertos fármacos alcalinizando o acidifi cando la orina.
Los cambios en la eliminación del fármaco al modificar el
pH urinario dependen del grado y la duración del cambio
en el pH y de la contribución de la reabsorción pasiva dependiente de pH a la eliminación total del fármaco. Este
efecto es mayor para los ácidos y bases débiles con un pKa
dentro de los límites del pH urinario (5 a 8). No obstante,
la alcalinización de la orina puede aumentar entre cuatro
y seis veces la excreción de un ácido relativamente fuerte,
como el salicilato, cuando se cambia el pH urinario de 6.4
a 8.0 y la fracción de fármaco no ionizado se reduce de 1 a
0.04 por ciento.
Excreción biliar y fecal. La membrana canalicular del hepatocito también posee transportadores análogos a los del riñón y éstos secretan de
manera activa fármacos y metabolitos hacia la bilis. La P-gp transporta
a una gran variedad de fármacos liposolubles anfi páticos, mientras que
MRP2 participa sobre todo en la secreción de los metabolitos conjugados de los fármacos (p. ej., conjugados de glutatión, glucurónidos y
algunos sulfatos). Por último, los fármacos y metabolitos presentes en la
bilis se expulsan hacia el aparato digestivo durante la digestión. Los enterocitos también expresan transportadores secretores en su membrana
apical, de manera que hay secreción directa de fármacos y metabolitos
desde la circulación general hasta la luz intestinal. Más adelante los fármacos y metabolitos pueden reabsorberse a partir del intestino, pero la
microflora intestinal deberá realizar su hidrólisis enzimática en algunos
casos, como sucede con los metabolitos conjugados, como los glucurónidos. Este reciclaje enterohepático, cuando es extenso, prolonga la
presencia del fármaco (o toxina) y sus efectos dentro del organismo
antes de su eliminación por otras vías. Es por esta razón que se pueden
administrar fármacos por vía oral para fijar sustancias excretadas en la
bilis. Por ejemplo, en el caso de la intoxicación por mercurio, se puede
administrar una resina por vía oral que se une al dimetilmercurio que se
excreta en la bilis, con lo que se previene su reabsorción y una mayor
toxicidad. El reciclaje enterohepático también tiene sus ventajas en la
creación de los fármacos. El ezetimibe es el primero de una clase nueva
de fármacos que reducen de manera específica la absorción intestinal de
colesterol (Lipka, 2003). Este fármaco se absorbe en la célula epitelial intestinal y se cree que interfi ere con el sistema transportador de
esteroles. De esta manera, impide el transporte de colesterol libre y
esteroles vegetales (fi toesteroles) desde la luz intestinal hasta la célula.
Este medicamento se absorbe con rapidez y es glucuronizado en la
célula intestinal antes de ser secretado hacia la sangre. El hígado absorbe con avidez ezetimibe desde la sangre porta y lo excreta hacia la
bilis, con lo que su concentración en la sangre periférica es reducida.
El conjugado con glucurónido es hidrolizado y absorbido, y es igual de
efectivo para inhibir la absorción de esterol. Este reciclaje enterohepá-
tico provoca que su semivida en el organismo sea mayor de 20 h. El
principal beneficio es que se reducen las lipoproteínas de baja densidad
(véase cap. 35).
Excreción por otras vías. La excreción de fármacos a través del sudor, la saliva y las lágrimas es insignifi cante desde el punto de vista
cuantitativo. La eliminación por tales vías depende de la difusión de
los medicamentos no ionizados liposolubles a través de las células epiteliales de las glándulas y depende del pH. Los fármacos excretados en
la saliva penetran en la cavidad bucal y desde ella son deglutidos. La
concentración de algunos de ellos en la saliva corresponde a la observada en plasma. Por tal razón, la saliva quizá sea un líquido biológico
útil para medir las concentraciones de medicamentos en situaciones en
que es difícil o incómodo obtener sangre. Los mismos principios son
válidos para la excreción de medicamentos en la leche materna. Esta
leche es más ácida que el plasma, razón por la cual los compuestos
alcalinos pueden hallarse un poco más concentrados en ella, y la concentración de compuestos ácidos en la leche es menor que en el plasma. Las sustancias no electrolíticas, como el etanol y la urea, llegan
con facilidad a la leche materna y alcanzan la misma concentración
que tienen en el plasma, independientemente del pH de la leche. Por
lo tanto, al administrar medicamentos a una mujer que se encuentra
alimentando a su hijo al seno materno es importante tener en mente
que el lactante tendrá contacto, hasta cierto grado, con el medicamento
o sus metabolitos. En algunos casos, durante el tratamiento con el bloqueador β atenolol, el lactante tendrá contacto con una gran cantidad
de fármaco (Ito y Lee, 2003). Si bien la excreción en el pelo y la piel
es insignifi cante, existen métodos sensibles para detectar cantidades
ínfimas de estos fármacos en estos tejidos y que tienen importancia
para la medicina forense.
METABOLISMO DE FÁRMACOS
Las características lipófi las que facilitan el paso de los medicamentos por las membranas biológicas y el acceso ulterior
al sitio de acción, obstaculizan su eliminación del organismo.
La excreción del fármaco intacto (sin cambios) a través de
los riñones interviene muy poco en la eliminación global
de casi todos los agentes terapéuticos, porque los productos
lipófilos que son fi ltrados por el glomérulo se resorben en
gran medida y se devuelven a la circulación general durante su paso por los túbulos renales. Por ello, el metabolismo
de fármacos y otros productos xenobióticos en metabolitos
más hidrófi los resulta esencial para la eliminación de tales
compuestos del organismo y la terminación de su actividad
biológica. Desde una perspectiva general, las reacciones de
biotransformación generan metabolitos inactivos más polares, que se excretan con facilidad al exterior. Sin embargo,
en algunos casos se producen metabolitos con potente actividad biológica o con propiedades tóxicas. Muchos de los
sistemas enzimáticos que transforman a los fármacos en sus
metabolitos inactivos también generan metabolitos con actividad biológica de compuestos endógenos, como sucede en
la biosíntesis de los esteroides.
El metabolismo de los fármacos o reacciones de biotransformación
se clasifi can como reacciones de funcionalización de la fase I o reacciones biosintéticas de la fase II (conjugación). Las reacciones de la
fase I presentan o exponen al grupo funcional del compuesto original,
como sucede en la hidrólisis; por lo general provocan la pérdida de la
actividad farmacológica, aunque existen ejemplos donde se retiene o
intensifica. En algunos casos raros, el metabolismo se acompaña de
una actividad farmacológica alterada. Los profármacos son los compuestos inactivos desde el punto de vista farmacológico creados para
aumentar las especies activas que alcanzan su sitio de acción. Los profármacos inactivos son convertidos rápidamente en metabolitos con
actividad biológica a menudo por la hidrólisis de un éster o un enlace
de amida. Éste es el caso de varios inhibidores de la enzima convertidora de angiotensina (angiotensin-converting enzyme, ACE) utilizados en el tratamiento de la hipertensión. Por ejemplo, el enalaprilo es
relativamente inactivo hasta que es convertido por la actividad de la
enterasa en enalaprilato diácido. Si no son excretados con rapidez hacia la orina, los productos de las reacciones de biotransformación de la
fase I reaccionan con compuestos endógenos para formar un conjugado
altamente hidrosoluble.
Las reacciones de conjugación de la fase II culminan en la formación
de un enlace covalente entre un grupo funcional en el compuesto original, o metabolito de fase I, con los derivados de manera endógena: ácido
glucurónico, sulfato, glutatión, aminoácidos o acetato. Estos conjugados fuertemente polares suelen ser inactivos y se excretan con rapidez
por orina y heces. Un ejemplo de un conjugado activo es el metabolito
6-glucurónido de morfi na, un analgésico más potente que el compuesto original.
Los sistemas enzimáticos que participan en la biotransformación
de los fármacos se ubican sobre todo en el hígado, si bien cualquier
tejido que se examina posee cierta actividad metabólica. El aparato
digestivo, los riñones y los pulmones también tienen un potencial
metabólico notable. Después de administrar un fármaco por vía oral,
gran parte de la dosis sufre desactivación metabólica en el epitelio
intestinal o el hígado antes de llegar a la circulación general. El llamado metabolismo de primer paso limita signifi cativamente la disponibilidad oral de fármacos que son altamente metabolizados. La
mayor parte de la actividad metabólica de los fármacos en el interior
de la célula se realiza en el retículo endoplásmico liso y el citosol,
aunque también hay biotransformación de fármacos en las mitocondrias, la cubierta nuclear y la membrana plasmática. El sistema enzimático que participa en las reacciones de la fase I se ubica sobre
todo en el retículo endoplásmico, mientras que los sistemas enzimá-
ticos de conjugación de la fase II son básicamente citosólicos. Con
frecuencia, los fármacos sometidos a biotransformación a través de
una reacción de la fase I en el retículo endoplásmico son conjugados
en este mismo sitio o en la fracción citosólica de la misma célula en
forma secuencial. Los encargados de estas reacciones de biotransformación son las isoformas del citocromo P450 (cytochrome P450,
CYP) y varias transferasas. Estas familias de enzimas, las importan tes reacciones que catalizan y su participación en el metabolismo de
los fármacos y las respuestas farmacológicas adversas se describen
con detalle en el capítulo 3
FARMACOCINÉTICA CLÍNICA
El principio fundamental de la farmacocinética clínica es que
se cuenta con una relación entre los efectos farmacológicos
de un medicamento y su concentración asequible (p. ej., en
sangre o plasma). Esta relación se ha comprobado para muchos fármacos y es de gran utilidad en el tratamiento de los
pacientes. En el caso de algunos medicamentos no se ha observado una relación sencilla o clara entre el efecto farmacológico y la concentración plasmática, mientras que en el de
otros resulta poco práctico medir sistemáticamente la concentración como parte de la vigilancia terapéutica. En la mayor parte de los casos, como se muestra en la fi gura 1-1, la
concentración del fármaco en sus sitios de acción depende
de la concentración en la circulación general. El efecto farmacológico resultante puede ser el efecto clínico deseado,
un efecto tóxico o, en algunos casos, un efecto que no tiene
relación con la efi cacia terapéutica o los efectos secundarios.
La fi nalidad de la farmacocinética clínica es proporcionar
tanto una relación cuantitativa entre dosis y efecto como un
marco de referencia para interpretar la concentración de los
fármacos en los líquidos biológicos por el bien del paciente.
La importancia de la farmacocinética en la atención de los
pacientes radica en la posibilidad de mejor eficacia terapéutica y evitar los efectos indeseables, lo que se puede lograr si
se aplican sus principios al elegir y modifi car los esquemas
medicamentosos.
Las variables fi siológicas y fi siopatológicas que establecen los ajustes posológicos en cada paciente a menudo lo
hacen como resultado de los parámetros farmacocinéticos
modificados. Los cuatro parámetros principales que rigen la
disposición de los fármacos son la eliminación, que es una
medida de la efi cacia del organismo para eliminar el fármaco; el volumen de distribución, que es una medida del espacio disponible en el organismo para contener al fármaco; la
semivida de eliminación, que es una medida de la velocidad
con la que se expulsa el fármaco del organismo, y la biodisponibilidad, que es la fracción del fármaco que se absorbe
como tal hacia la circulación general.
Eliminación
Al concebir un esquema racional para administrar un medicamento durante un tiempo prolongado, el concepto más
importante que debe considerarse es la eliminación. Por lo
general, el médico desea mantener una concentración estable
del fármaco dentro de la ventana terapéutica asociado a una
eficacia terapéutica y mínima toxicidad. Suponiendo que la
biodisponibilidad sea completa, la concentración estable del
fármaco en el organismo se alcanza cuando la velocidad de
eliminación es igual a la velocidad de la administración. Por
lo tanto:
Dosificación = CL . Css (1-2)
donde CL es la eliminación (clearance) desde la circulación
sistémica, y Css, la concentración en estado estable (steadystate concentration) del fármaco. Si se conoce la concentración en estado estable buscada en plasma o sangre, la velocidad de eliminación del medicamento será el elemento que
rija la frecuencia con que debe administrarse.
El concepto de eliminación es de gran utilidad en el campo de la
farmacocinética clínica porque su valor para determinado fármaco suele ser constante en el rango de concentraciones que se observan en la
clínica. Una prueba de ello es que los sistemas para eliminar fármacos,
como enzimas metabolizantes y transportadores (véanse caps. 2 y 3) no
suelen saturarse y, por lo tanto, la velocidad absoluta de eliminación del
fármaco es básicamente una función lineal de su concentración plasmá-
tica. Esto es, la mayor parte de los fármacos se elimina siguiendo una
cinética de primer orden, donde una fracción constante del fármaco en
el organismo se elimina por unidad de tiempo. Cuando los mecanismos
de eliminación se saturan, la cinética se acerca al orden de cero, donde
una cantidad constante del fármaco se elimina por unidad de tiempo. En
este caso, la eliminación (CL) varía según la concentración del fármaco,
a menudo de acuerdo con la ecuación siguiente:
CL = vm/(Km 1 C) (1-3)
donde K
m representa la concentración en la cual se alcanza la mitad de
la velocidad máxima de eliminación (en unidades de masa/volumen)
y vm es igual a la velocidad máxima de eliminación (en unidades de
masa/tiempo). Así, la eliminación se calcula en unidades de volumen/
tiempo. La ecuación anterior es análoga a la de Michaelis-Menten, vá-
lida para la cinética de enzimas. Preparar programas posológicos para
los fármacos en cuestión es más complejo que cuando la eliminación
es de primer orden y la “eliminación” es independiente de la concentración del producto medicamentoso (véase más adelante en este capítulo).
Los principios de la eliminación de medicamentos son semejantes
a los de la fi siología renal; por ejemplo, la eliminación de creatinina
se defi ne como la velocidad de eliminación de dicho metabolito en la
orina, en relación con su concentración en plasma. En su grado más
sencillo, la eliminación de un producto medicamentoso es la velocidad de eliminación por todas las vías, normalizada a la concentración
del fármaco C en algún líquido biológico en que pueda efectuarse la
medición:
CL = velocidad de eliminación/C (1-4)
De esta manera, cuando la eliminación es constante, la velocidad con que se elimina el fármaco es directamente proporcional a su
concentración. Es importante recordar que la eliminación no indica
la cantidad de fármaco eliminado, sino el volumen de líquido biológico,
como sangre o plasma, del cual se tendría que eliminar por completo el fármaco para explicar la eliminación (p. ej., ml/min/kg). Así,
la eliminación puede definirse como eliminación sanguínea (blood
clearance, CLb), eliminación plasmática (plasma clearance, CLp) o
eliminación basada en la concentración de fármaco libre (clearance
unbound drug, CLu), dependiendo de la medida que se toma en cuenta
(Cb, Cp o Cu).
La eliminación por los órganos encargados de ella es aditiva. La
eliminación del fármaco puede ser consecuencia de mecanismos que
tienen lugar en riñones, hígado y otros órganos. Si la velocidad de eliminación correspondiente a un órgano dado se divide entre la concentración del fármaco (p. ej., la concentración plasmática), se obtendrá
la depuración particular de ese órgano. En conjunto, al sumarse, estas
depuraciones separadas equivaldrán a la eliminación sistémica total:
CLrenal + CLhepática + CLotras + CL (1-5)
Otras vías de eliminación son la saliva o el sudor, la secreción hacia
el aparato digestivo, la eliminación volátil a partir del pulmón y el metabolismo en otros sitios como la piel.
La eliminación generalizada puede valorarse en una situación de
equilibrio basal conforme a la ecuación 1-2. En lo que se refi ere a una
sola dosis de medicamento con biodisponibilidad completa y cinética
de eliminación de primer orden, la eliminación generalizada (sistémica)
puede calcularse con base en el equilibrio de masas y la integración de
la ecuación 1-4 en función del tiempo:
CL = dosis/AUC (1-6)
donde AUC es el área total debajo de la curva (area under the curve),
que describe la concentración del fármaco en la circulación general en
función del tiempo (desde cero hasta infi nito), como en la figura 1-5.
Ejemplos. En el Apéndice II se señala que la eliminación de la cefalexina del plasma es de 4.3 ml/min/kg y por la orina se excreta 90%
del fármaco intacto. Si se considera el caso de un varón de 70 kg de
peso, la eliminación desde el plasma equivaldría a 301 ml/min, y la
eliminación por riñones comprendería 90% de la eliminación. En otras
palabras, los riñones pueden excretar cefalexina a un ritmo tal que ésta
se elimina por completo a razón de aproximadamente 270 ml de plasma
por minuto (eliminación renal 5 90% de la eliminación total). Dado que
la eliminación se supone constante en el paciente estable, la velocidad
total de eliminación de la cefalexina dependerá de la concentración del
fármaco en el plasma (ecuación 1-4).
El antagonista de receptores adrenérgicos β propranolol se depura
desde la sangre a razón de 16 ml/min/kg (en un varón de 70 kg de peso
serían 1 120 ml/min), y tal tarea la realiza casi exclusivamente el hígado. Por lo tanto, este último puede extraer todo el fármaco contenido en
1 120 ml de sangre cada minuto. A pesar de que el hígado es el órgano
“dominante” de eliminación, la desaparición de algunos medicamentos
desde el plasma rebasa el índice o velocidad de fl ujo de plasma y sangre
que llega al órgano. A menudo tal situación acaece porque el medicamento muestra fácilmente partición entre los eritrocitos (red blood cells,
RBC), y la cantidad de fármaco que llega al órgano de excreción es
muchísimo mayor que la que se esperaría por la medición de su concentración en plasma. La relación entre la eliminación plasmática (p) y la
sanguínea (blood, b) en estado estable la da la ecuación siguiente:
Puede calcularse la “salida” del medicamento desde la sangre, al dividir la eliminación desde el plasma, entre la razón de concentraciones sangre/plasma del medicamento, obtenida por conocimiento del hematócrito
(H = 0.45) y la razón de concentraciones eritrocíticas/plasmáticas. En casi todos los casos, la “extracción” o salida desde la sangre será menor
que el fl ujo sanguíneo por el hígado (1.5 L/min), o si participa también la
excreción por riñones, la suma de las corrientes sanguíneas de ambos órganos de eliminación. Por ejemplo, la desaparición del tracolim desde el
plasma, que es de casi 2 L/min, tiene una magnitud dos veces mayor que
la velocidad de fl ujo plasmático al hígado e incluso rebasa la corriente
sanguínea de dicho órgano, a pesar de que el órgano en cuestión es el sitio
predominante del metabolismo extenso de dicho fármaco. Sin embargo,
después de tomar en consideración la distribución extensa de tacrolimús en
los eritrocitos, la “salida” de él desde la sangre es sólo de 63 ml/min, y
en realidad se trata de un medicamento con eliminación baja y no alta,
como cabría interpretar si se considera la cifra de eliminación o desaparición del plasma. En ocasiones, la desaparición desde la sangre por
el metabolismo rebasa la corriente sanguínea por el hígado y ello denota
metabolismo extrahepático, En el caso del antagonista de receptores β1 esmolol (11.9 L/min), la cifra de eliminación desde la sangre es mayor que
el gasto cardíaco (aproximadamente 5.3 L/min), porque el fármaco se metaboliza de manera efi caz con las esterasas que están en los eritrocitos.
Para entender los efectos de variables patológicas y fisiológicas en
la eliminación de los medicamentos, en particular la correspondiente
a un órgano determinado, será útil defi nir con mayor amplitud la eliminación. La velocidad de “presentación” del medicamento al órgano
sería el producto del flujo sanguíneo (Q) por la concentración del medicamento en sangre arterial (CA), y la velocidad con que el fármaco
sale del órgano sería el producto de dicho fl ujo por la concentración
del medicamento en sangre venosa (CV). La diferencia entre estas velocidades, en equilibrio dinámico o estado estable, sería la velocidad de
eliminación del fármaco:

La expresión (CA - CV)/CA en la ecuación 1-9 se puede calificar
como la razón de extracción (E) del fármaco. Aunque no se utiliza en la
práctica médica general, resulta útil calcular la razón de extracción del
fármaco para modelar los efectos que tiene el trastorno de cierto órgano
metabolizante sobre la eliminación y para diseñar las propiedades terapéuticas ideales de los fármacos que se están elaborando.
Eliminación por el hígado. Los conceptos planteados en la ecuación 1-9
tienen enorme trascendencia en lo que toca a medicamentos que son eliminados por el hígado. Considérese el caso de un producto farmacéutico
que se elimina con eficacia de la sangre mediante procesos hepáticos,
es decir, metabolismo o excreción, o ambos, del medicamento intacto
en la bilis. En este caso, será pequeña la concentración del fármaco en
la sangre que salga del hígado, la razón de extracción se acercará a la
unidad y la eliminación del medicamento de la sangre tendrá como elemento limitante el flujo de este líquido por el hígado. Los fármacos que
son eliminados eficazmente por esta víscera (p. ej., los señalados en el
Apéndice II, cuyas velocidades de eliminación sistémica exceden de
6 ml/min/kg, como diltiazem, imipramina, lidocaína, morfina y propranolol), muestran restricción en su velocidad de eliminación, no por procesos intrahepáticos, sino por la rapidez con que son transportados por la
sangre a los sitios de eliminación presentes en el hígado.
Quedan por considerar algunos otros aspectos complejos. Por ejemplo, las ecuaciones expuestas en párrafos anteriores no tienen en cuenta
la unión del fármaco a componentes de la sangre y los tejidos, ni permiten calcular la habilidad intrínseca del hígado para eliminar un medicamento en caso de no haber las limitaciones impuestas por el flujo
de sangre, lo cual se ha llamado eliminación intrínseca. En términos
bioquímicos y bajo circunstancias de primer orden, la eliminación intrínseca es una medida de la proporción de los parámetros cinéticos de
Michaelis-Menten para el proceso de eliminación (p. ej., vm/Km) y, por
lo tanto, refl eja la capacidad metabólica máxima o de transporte del
órgano que realiza la eliminación. Se han propuesto varias extensiones de las relaciones de la ecuación 1-9 para incluir expresiones para
la unión a proteínas y la eliminación intrínseca en varios modelos de
eliminación hepática (Kwon y Morris, 1997). Todos estos modelos indican que cuando la capacidad del órgano eliminador para metabolizar el
fármaco es considerable comparada con la velocidad con que el fármaco es presentado al mismo, la eliminación será similar a la irrigación del
órgano. Por el contrario, cuando la capacidad metabolizante es pequeña
comparada con la velocidad con que se presenta el fármaco, la eliminación será directamente proporcional a la fracción de fármaco libre en la
sangre y a la eliminación intrínseca del mismo. Si se conocen estos conceptos es posible comprender una serie de resultados experimentales
que quizá son dudosos. Por ejemplo, la inducción enzimática o una hepatopatía pueden alterar la velocidad con que se metaboliza el fármaco
en determinado sistema enzimático microsomal hepático, sin cambiar la
eliminación global en todo el animal. Para un fármaco con una razón de
extracción alta, la irrigación restringe la eliminación, y los cambios en
la eliminación intrínseca por inducción enzimática o hepatopatía deben
tener muy pocos efectos. Asimismo, para fármacos con una razón de extracción alta, los cambios en la unión a proteínas por alguna enfermedad
o por competencia de otros fármacos deben tener muy poco efecto sobre
la eliminación. Por el contrario, los cambios en la eliminación intrínseca
y la unión a proteínas modifi can la eliminación de los fármacos con una
eliminación intrínseca reducida, como la warfarina y, por lo tanto, las
razones de extracción; sin embargo, los cambios de la irrigación tendrán
muy pocos efectos.
Eliminación renal. La eliminación renal de un fármaco provoca su
aparición en la orina. Al valorar los efectos que tiene una nefropatía
sobre la eliminación de un fármaco es importante tomar en cuenta las
complicaciones ligadas a la fi ltración, la secreción activa en el túbulo
renal y la reabsorción, además del fl ujo. La velocidad de filtración de un
medicamento depende del volumen del líquido fi ltrado por el glomérulo
y la concentración libre del fármaco en plasma, dado que no se filtra el
que está unido a proteínas. La velocidad de secreción del medicamento
por el riñón dependerá de la “eliminación” intrínseca del fármaco por
acción de los transportadores que intervienen en la secreción activa, modificada por la unión del medicamento a proteínas plasmáticas, el grado de
saturación de dichos transportadores, y la rapidez de llegada del medicamento al sitio secretor. Además, habrá que considerar los procesos
que intervienen en la resorción del fármaco desde el líquido tubular. Las
influencias de los cambios en la unión a proteínas, el fl ujo sanguíneo y el
número de nefronas funcionales son análogas a los principios expuestos
en párrafos anteriores en lo referente a la eliminación por el hígado.
DISTRIBUCIÓN
Volumen de distribución. El segundo parámetro fundamental que resulta útil para entender los mecanismos de eliminación de un fármaco es el volumen. El volumen de distribución
(V) relaciona la cantidad de medicamento en el organismo
con la concentración que tiene (C) en sangre o plasma, según
el líquido que se mida. Dicho volumen no necesariamente se
refiere a un volumen fisiológico identificable, sino sólo al volumen de líquido que se requeriría para contener todo el fármaco en el cuerpo a las mismas concentraciones en que está
presente en sangre o plasma:
Cantidad de fármaco en el organismo/V 5 C o (1-10)
V 5 cantidad de fármaco en el organismo/C
Por lo tanto, el volumen de distribución de un fármaco
refleja su presencia en los tejidos extravasculares y no en el
plasma. El volumen plasmático de un hombre de 70 kg es
de 3 L, su volumen sanguíneo es de 5.5 L, su volumen de
líquido extracelular excluyendo al plasma es de 12 L y su
volumen de agua total es de aproximadamente 42 litros.
Muchos fármacos poseen volúmenes de distribución mucho mayores que estas cifras. Por ejemplo, si hubiera 500 µg del glucósido
cardíaco digoxina en el cuerpo de un individuo de 70 kg, se observaría
una concentración plasmática aproximada de 0.75 ng/ml. Si se divide la
cantidad de fármaco en el organismo entre su concentración plasmática
se obtiene un volumen de distribución para la digoxina de alrededor de
667 L, o un valor cerca de 10 veces mayor que el volumen total de un
hombre de 70 kg. De hecho, la digoxina se distribuye de preferencia en
el músculo y el tejido adiposo y en sus receptores específicos (NA1,
K1-ATPasa), lo que deja una cantidad muy pequeña de fármaco en el
plasma. En cuanto a los fármacos que se fi jan de manera extensa a las
proteínas plasmáticas pero no se fi jan a los componentes hísticos, el volumen de distribución será similar al del volumen plasmático, ya que el
medicamento unido a las proteínas se puede medir en la mayor parte de
los análisis farmacológicos. Por el contrario, algunos fármacos tienen
un volumen de distribución alto aunque la mayor parte de la sustancia
en la circulación se encuentre unida a la albúmina, ya que estos fármacos también se secuestran en otros sitios.
El volumen de distribución varía considerablemente según el grado
relativo de unión de alta afi nidad con otros sitios receptores, proteínas
plasmáticas e hísticas, el coefi ciente de fragmentación del fármaco en
la grasa y la acumulación en los tejidos poco irrigados. Como es de
esperarse, el volumen de distribución de los fármacos difi ere según la
edad del paciente, el sexo, la composición corporal y la presencia de
alguna enfermedad. El agua total en los lactantes menores de un año
de edad, por ejemplo, es de 75 a 80% del peso corporal, mientras que
en un adulto de sexo masculino es de 60% y en uno de sexo femenino es
de 55 por ciento.
Para describir la distribución de los medicamentos se emplean algunos términos volumétricos que se han obtenido o calculado de diversos
modos. El volumen de distribución que se defi ne en la ecuación 1-10
considera al organismo como un solo compartimiento homogéneo. En
ese modelo unicompartimental, todo el fármaco que llegue al organismo pasa directamente al compartimiento central, y la distribución de
la sustancia es instantánea en todo el volumen (V). La eliminación o
eliminación desde dicho compartimiento sigue una cinética de primer
orden, como se defi ne en la ecuación 1-4, es decir, la cantidad de fármaco eliminado por unidad de tiempo depende de la cantidad (concentración) del producto medicamentoso en el compartimiento corporal.
En la figura 1-3A y la ecuación 1-11 se describe la disminución de la
concentración plasmática con el paso del tiempo, correspondiente a un
medicamento introducido en dicho compartimiento:
C = (dosis/V) . exp(-kt) (1-11)
donde k es la constante de rapidez de eliminación que expresa la fracción del fármaco “extraído” o eliminado desde el compartimiento, por
unidad de tiempo. Dicha constante guarda relación inversa con el periodo de semieliminación, o semivida, del medicamento (k 5 0.693/t 1
2
--).
El modelo unicompartimental “ideal” que se ha señalado, no describe toda la evolución cronológica que sigue la concentración plasmá-
tica; es decir, habrá que diferenciar entre algunos depósitos tisulares y
el compartimiento central, y la concentración del medicamento parece
disminuir de una manera que podría describirse en términos exponenciales múltiples (fig. 1-3B). Sin embargo, el modelo unicompartimental
es sufi ciente para aplicarse en casi todas las situaciones clínicas y a casi
todos los medicamentos. Efectivamente, el hecho de conocer la vida del
fármaco en el compartimiento central tiene un efecto directo y significativo sobre el intervalo posológico apropiado de administración.
Velocidad de distribución. La caída exponencial que se observa en
los fármacos que son eliminados del organismo por medio de una cinética de primer orden es consecuencia de las diferencias en la velocidad con la que el fármaco se equilibra con los tejidos y dentro de
los mismos. La velocidad de equilibrio depende de la relación entre la
perfusión del tejido y la fragmentación del fármaco en el tejido. En muchos casos, varios grupos de tejidos con una relación entre perfusión y
fragmentación similar se equilibran básicamente a la misma velocidad,
de manera que sólo se observa una fase de distribución (descenso inicial
rápido de la concentración del fármaco inyectado por vía intravenosa,
como se muestra en la fig. 1-3B). Es como si el fármaco comenzara en
un volumen “central” (fi g. 1-1), que consta de un depósito plasmático
y otro hístico que se equilibra rápidamente con el medicamento distribuyéndolo hasta lograr un volumen “fi nal”, en cuyo punto las concentraciones plasmáticas disminuyen en forma logarítmica lineal a una
velocidad constante de k (fig. 1-3B). Una manera de mirar el modelo de
compartimientos múltiples para la disposición de un fármaco es como
si la sangre y los órganos magros con una perfusión abundante como el
corazón, cerebro, hígado, pulmón y riñones se agruparan formando un
solo compartimiento central, mientras que los tejidos menos irrigados,
como músculo, piel, grasa y hueso, actúan como el compartimiento final
(esto es, el compartimiento hístico).
Si el modelo o la proporción de fl ujos de sangre hacia diversos tejidos cambia dentro de una persona o difi ere entre uno y otro individuos,
también se modifi carán las velocidades de distribución del medicamento en los tejidos. Sin embargo, los cambios en el flujo sanguíneo
también pueden hacer que algunos tejidos que estaban al principio en
el volumen “central” se equilibren con una lentitud mucho mayor, al
grado de que aparezcan sólo en el volumen “fi nal”; esto significa que
parecerá que los volúmenes centrales varían con estados patológicos
que alteran el flujo de sangre regional (como se observaría en la cirrosis hepática). Después de la administración intravenosa rápida, las
concentraciones del fármaco en plasma pueden ser mayores en sujetos
con riego defi ciente (como sería en el choque), que si el riego sanguíneo
fuera más adecuado. Estas altas concentraciones sistémicas ocasionan
a su vez concentraciones más altas (y efectos más intensos) en tejidos
como los de encéfalo y corazón, cuyo gran riego no ha sido disminuido
por la alteración del estado hemodinámico. Por lo tanto, el efecto de un
medicamento en diversos sitios de acción puede variar de acuerdo con
el riego sanguíneo que reciben.
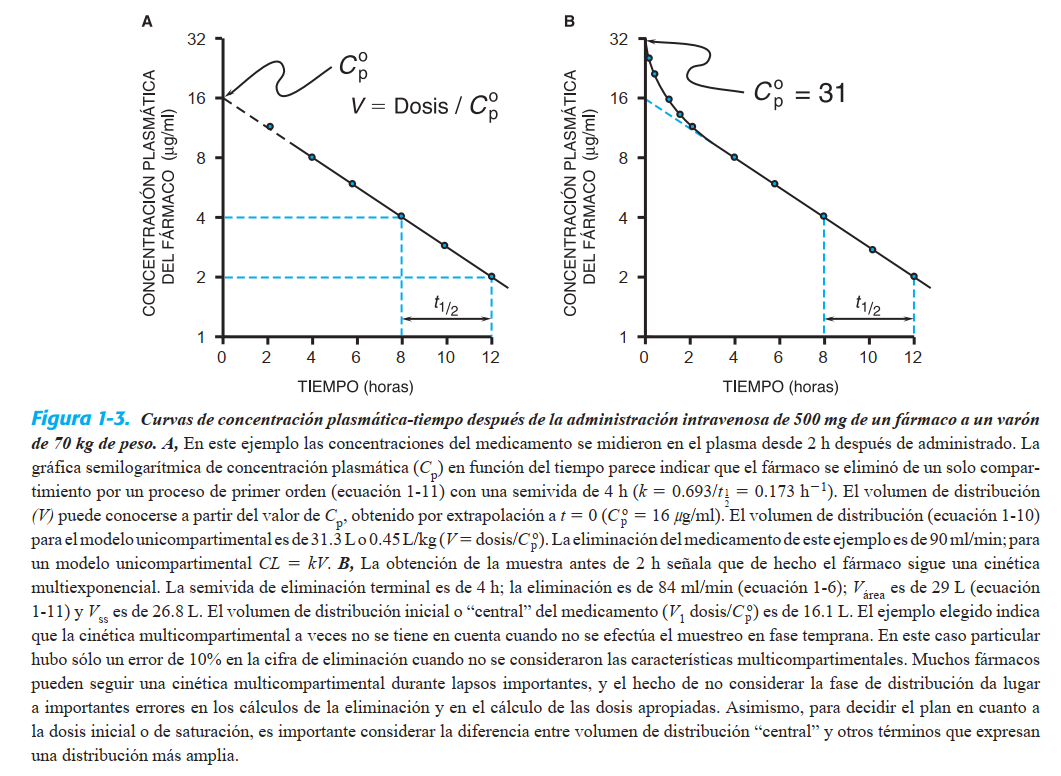
Volumen multicompartimental. Se han utilizado dos términos diferentes para describir el volumen de distribución de fármacos que siguen
una desintegración exponencial múltiple. El primero, llamado Várea, se
calcula como la razón aritmética entre la eliminación y la rapidez de
disminución de la concentración durante la fase de eliminación (final)
de la curva de concentración logarítmica, en función del tiempo:

El cálculo del parámetro anterior es directo y permite conocer
el volumen después de administrar una sola dosis del medicamento
por vía intravenosa u oral (donde la dosis debe ser corregida por la biodisponibilidad). Sin embargo, otro volumen de distribución multicompartimental puede ser más útil, en especial cuando interesa conocer el efecto
de los estados patológicos sobre la farmacocinética. El volumen de distribución en estado estable (volume of distribution at steady state, Vss)
es aquel en que el fármaco se distribuiría durante el estado de equilibrio
si se hallara en todo ese volumen en la misma concentración en que está
en el líquido donde se mide (plasma o sangre). Vss también se puede
apreciar como se muestra en la ecuación 1-13, donde VC es el volumen
de distribución del fármaco en el compartimiento central y VT es el mismo volumen en el compartimiento hístico (tisular):

A pesar de que Várea es un parámetro cómodo y de cálculo fácil
varía cuando la constante de velocidad de eliminación del medicamento
cambia, aun sin modifi cación del espacio para distribución. Lo anterior
se debe a que la velocidad terminal de disminución de la concentración
del fármaco en sangre y plasma depende no sólo de la eliminación, sino
de las velocidades de distribución del producto entre los volúmenes
“central” y “fi nal”. El Vss no tiene esta desventaja. Cuando se aplica la
farmacocinética para decidir la posología, las diferencias entre Várea y
V
ss por lo regular carecen de importancia clínica. No obstante, ambas
se incluyen en el cuadro de Datos farmacocinéticos, del Apéndice II,
según aparecen en las publicaciones sobre el tema.
Semivida
La semivida (t 1)
2
-- es el tiempo que necesita la concentración
plasmática o la cantidad del medicamento en el cuerpo para
disminuir a la mitad. En el caso más sencillo, que es el del
modelo unicompartimental (fig. 1-3A), la semivida puede
calcularse fácilmente y utilizarse para tomar decisiones en
cuanto a la dosifi cación del medicamento (posología). Sin
embargo, como se señala en la figura 1-3B, las concentraciones del fármaco en plasma suelen seguir un modelo de
disminución multiexponencial, lo cual hace posible calcular
dos o más términos para la semivida.
En lo pasado, los valores de semivida se informaban en términos
de la fase de eliminación logarítmica lineal. Sin embargo, conforme
se logró mayor sensibilidad analítica, las concentraciones menores
medidas parecían producir valores crecientes de semivida terminal.
Por ejemplo, se ha observado una semivida terminal de 53 h en el
caso de la gentamicina (en comparación con las 2 a 3 h señaladas en
el Apéndice II, las cuales son más relevantes desde el punto de vista clínico), y el ciclo por la bilis quizás explique el valor terminal de
120 h de la indometacina (en comparación con la semivida de 24 h
señalada en el Apéndice II). La semivida terminal prolongada de algunos medicamentos es consecuencia de su acumulación en los tejidos
por su administración crónica o durante periodos más cortos de tratamiento
pero con una dosis alta. Éste es el caso de la gentamicina, cuya semivida
terminal es nefrotóxica y ototóxica. La importancia de una vida media particular puede defi nirse en términos de la fracción de la eliminación y
el volumen de distribución que se relacionan con cada semivida, y de
si las concentraciones plasmáticas o las cantidades del medicamento
en el cuerpo se relacionan mejor con las respuestas medidas. Las cifras únicas de semivida señaladas para cada fármaco en el Apéndice II expresan el periodo de semieliminación o semivida de mayor interés clínico.
En los estudios sobre las propiedades farmacocinéticas de los fármacos durante la enfermedad, la semivida es un parámetro derivado que
cambia en función de la eliminación y el volumen de distribución. La
fórmula siguiente ofrece una relación aproximada y útil entre la semivida, la eliminación y el volumen de distribución:
t
1/2 -= 0.693 ? Vss/CL (1-14)
La eliminación es la medida de la capacidad que tiene el
organismo para eliminar el fármaco; así, al disminuir la capacidad de eliminación, como sería por alguna entidad patológica, cabría esperar que aumentara la semivida del medicamento en el organismo. No obstante, esta relación recíproca
es exacta sólo cuando la enfermedad no modifi ca el volumen
de distribución. Por ejemplo, la semivida del diazepam aumenta conforme lo hace la edad del individuo; sin embargo,
lo que cambia en función de la edad no es la eliminación,
sino el volumen de distribución. De igual modo, los cambios en la unión del fármaco a proteínas pueden alterar su
eliminación y también su volumen de distribución, y ocasionar cambios impredecibles en la semivida, en función de la
enfermedad. Por ejemplo, la semivida de la tolbutamida es
menor en sujetos con hepatitis viral aguda, exactamente lo
contrario de lo que se esperaría. La enfermedad modifica la
unión a proteínas en plasma y tejidos, de tal manera que, en
vez de que cambie el volumen de distribución, la eliminación
aumenta porque existen mayores concentraciones del medicamento libre en el torrente sanguíneo.
Aunque en ocasiones es un índice defi ciente de la eliminación de un fármaco del organismo por sí mismo (el fármaco
desaparece en ocasiones por la formación de metabolitos no
detectados pero que tienen efectos terapéuticos o indeseables), la semivida indica el tiempo necesario para alcanzar
el estado estable después de iniciar o cambiar un esquema
posológico (p. ej., cuatro semividas hasta alcanzar aproximadamente 94% de un estado estable nuevo) y el tiempo
que tarda un fármaco en eliminarse del organismo, además
de constituir una medida para calcular el intervalo adecuado para administrar cada dosis (véase más adelante en este
capítulo)
Estado estable. La ecuación 1-2 (velocidad de administración = CL . C
ss) indica que la concentración estable finalmente se obtiene cuando el fármaco se administra a una
velocidad constante. En este punto, la eliminación del fármaco (producto de eliminación y concentración; la ecuación
1-4) será igual al índice de disponibilidad del fármaco. Este
concepto se extiende también a la administración regular e intermitente (p. ej., 250 mg del fármaco cada 8 h). Durante
cada intervalo, la concentración del fármaco se incrementa
con la absorción y desciende con la eliminación. Durante el
estado estable, el ciclo se repite de manera idéntica en cada
intervalo (véase fig. 1-4). La ecuación 1-2 sigue aplicándose
para la administración intermitente, pero ahora describe la
concentración estable promedio (Css) durante el intervalo entre dos dosis.
Grado y tasa de biodisponibilidad
Biodisponibilidad. Es importante diferenciar entre la tasa
(velocidad) y el grado de absorción de un medicamento, y la
cantidad de fármaco que llega al fi nal a la circulación general.
La cantidad de medicamento que llega a la circulación general depende no sólo de la dosis administrada, sino también
de la fracción de la dosis (F) que es absorbida y que escapa de
la eliminación de primer paso. Esta fracción es la biodisponibilidad del fármaco. Ya antes se señalaron las causas de la
absorción incompleta; también, como se indicó, si el medicamento se metaboliza en el epitelio intestinal o el hígado, o se
excreta en la bilis, parte del fármaco activo absorbido en las
vías gastrointestinales terminará por ser eliminado antes de
que llegue a la circulación general y se distribuya hacia los
sitios de acción.
Si se conoce la razón de extracción (EH) del medicamento al pasar
por el hígado (ecuación 1-9), es posible conocer la máxima disponibilidad que habrá de él después de ingerido (Fmáx), en el supuesto de que la
eliminación hepática siga una cinética de primer orden:
F máx = 1 - EH = 1 - (CLhepático/Qhepático) (1-15)
Con base en la ecuación anterior, si la eliminación del fármaco
en la sangre que llega al hígado es grande en relación con el flujo
a dicha víscera, será pequeña la disponibilidad después de la ingestión
del producto (p. ej., lidocaína o propranolol); esta disminución de la
disponibilidad está en función del sitio fisiológico desde el cual se
absorbe el medicamento, y ninguna modifi cación en la presentación
mejorará la disponibilidad en una situación de cinética lineal. La absorción incompleta o el metabolismo intestinal (o ambos) incompletos
después de la ingestión del producto, en la práctica reducirá dicho valor máximo previsible de F.
Si un medicamento se suministra por una vía en que se produzca
pérdida de primer paso, las ecuaciones presentadas que contienen los
términos dosis o dosifi cación (ecuaciones 1-2, 1-6, 1-11 y 1-12) también deben incluir el término F de biodisponibilidad, de modo que se
utilice la dosis o dosifi cación disponible del producto. Por ejemplo, la
ecuación 1-2 se modifi ca hasta obtener:
F . dosificación = CL . Css (1-16)
donde el valor de F es de 0 a 1. El valor de F varía ampliamente en
los fármacos que se administran por vía oral. El etidronato, que es un
bisfosfonato utilizado para estabilizar la matriz ósea en el tratamiento
de la enfermedad de Paget y la osteoporosis, tiene un F de 0.03, lo
que signifi ca que sólo 3% del medicamento aparece en el torrente sanguíneo después de su administración oral. En el caso del etidronato,
el tratamiento por vía oral sigue siendo de utilidad y la dosis que se
administra por kilogramo es mayor que la que se utilizaría por vía intravenosa.
Velocidad de absorción. En general, la velocidad de absorción de un fármaco no influye en la concentración promedio en estado estable en que se halla en el plasma, pero aun
así infl uye en la farmacoterapia. Si el producto se absorbe con
gran rapidez (p. ej., la dosis que se aplica por vía intravenosa
rápida) y tiene un volumen “central” pequeño, la concentración del medicamento será grande en un principio, después
de lo cual disminuirá a medida que el medicamento se distribuya hasta alcanzar su volumen “fi nal” (mayor) (fig. 1-3B).
Cuando el mismo fármaco se absorbe con mayor lentitud
(p. ej., por goteo lento) se distribuirá durante el lapso de su administración, y las concentraciones máximas serán menores y
surgirán más tarde. Los preparados de liberación controlada
se concibieron para que la absorción sea lenta y sostenida
y así producir una concentración plasmática/perfil cronoló-
gico menos fl uctuante durante el intervalo entre una y otra
dosis, en comparación con presentaciones de liberación más
inmediata. Un medicamento particular puede originar efectos
deseables e indeseables en diversos sitios del organismo, y la
rapidez de distribución en esos sitios quizá no sea la misma.
De ese modo, las intensidades relativas de dichos efectos de
un producto pueden variar transitoriamente cuando se cambia el ritmo de administración. Para obtener los efectos favorables de un fármaco es necesario conocer su concentración
plasmática ideal o deseable, y el resultado terapéutico mejora
si se mantienen esos límites y se evitan las variaciones pronunciadas entre las concentraciones máxima y mínima.
Farmacocinética no lineal
En farmacocinética, la falta de linealidad (es decir, cambios en parámetros como eliminación, volumen de distribución y semivida en función
de la dosis o la concentración) por lo regular depende de la saturación
de la unión a proteínas, el metabolismo por el hígado, o el transporte
activo del medicamento a los riñones.
Unión saturable a proteínas. Al aumentar la concentración molar de un
medicamento, la fracción libre acaba por incrementarse también (al saturarse todos los sitios de unión), si bien esto suele ocurrir sólo cuando
las concentraciones del producto farmacéutico en el plasma alcanzan
órdenes de decenas a centenas de microgramos por mililitro. Cuando
un medicamento es metabolizado por el hígado con una proporción de
eliminación intrínseca/extracción baja, la saturación de la unión a proteínas plasmáticas hará que V y la eliminación (CL) aumenten conforme
lo hagan las concentraciones del medicamento; por tanto, la semivida
puede permanecer constante (ecuación 1-14). En el caso de dicho medicamento, C
ss no se incrementará de manera lineal conforme lo haga
el ritmo de administración del producto. Si los agentes medicamentosos
son depurados con índices de eliminación intrínseca/extracción grandes, C
ss puede seguir siendo linealmente (directamente) proporcional
a la tasa de administración del fármaco. En tal caso, la eliminación por
el hígado no cambiará y el incremento de V aumentará la vida media
de desaparición, al disminuir la fracción del medicamento total en el
organismo que llega al hígado por unidad de tiempo. Casi todos los fármacos quedan entre los dos extremos mencionados, y es difícil predecir
los efectos de la unión no lineal a proteínas.
Metabolismo saturable. En esta situación, la ecuación de MichaelisMenten (ecuación 1-3) por lo regular describe la falta de linealidad.
Sin duda, todos los mecanismos activos son saturables, pero parecerán
lineales si las cifras de las concentraciones medicamentosas observadas
en la práctica son mucho menores que Km. Cuando la concentración de
un fármaco es mayor que Km, se observa una cinética no lineal. Las consecuencias principales de la saturación del metabolismo o el transporte
son opuestas a las que se observan al saturarse la unión a proteínas. Esta
última origina incremento de CL, puesto que éste aumenta con la concentración del fármaco, mientras que la saturación del metabolismo o el
transporte reduce a CL. Cuando ambas circunstancias son simultáneas
prácticamente anulan los efectos de la otra y el resultado es una cinética
sorprendentemente lineal; por ejemplo, con el ácido salicílico esto sucede a lo largo de límites defi nidos de concentraciones.
El metabolismo saturable hace que el metabolismo de primer paso
oral sea menor de lo previsto (mayor F) y haya un incremento fraccionario mayor en Css que el incremento fraccionario correspondiente
en la velocidad de administración del fármaco. La situación anterior se
aprecia mejor si se sustituye la ecuación 1-3 en la ecuación 1-2 y se despeja la concentración en estado estable:

Conforme la dosifi cación se aproxima a la velocidad de eliminación
máxima, el denominador de la ecuación 1-17 se acerca a cero y Css aumenta de modo desproporcionado. Como la saturación del metabolismo no tiene efecto alguno en el volumen de distribución, la eliminación y la rapidez
relativa de la eliminación del medicamento disminuyen conforme aumenta
la concentración; por tal motivo, la curva de nivel/tiempo plasmática logarítmica C
p es cóncava y en fase de reducción hasta que el metabolismo
se torna sufi cientemente desaturado y aparece la eliminación de primer
orden. Por todo lo comentado, el concepto de semivida constante no es
aplicable al metabolismo no lineal que se observa en los límites habituales
de las concentraciones clínicas. Por consiguiente, es difícil e impredecible
cambiar el ritmo de dosis de un medicamento con metabolismo no lineal,
porque el estado estacionario resultante se alcanza de manera más lenta, y
como dato importante, el efecto no guarda proporción con la alteración del
ritmo posológico, es decir, la frecuencia de administración de dosis.
El anticonvulsivo fenitoína constituye un ejemplo de fármaco para el
cual el metabolismo se satura en el límite terapéutico de concentraciones
(véase Apéndice II) y la semivida varía de 7 a 42 h. Km (5 a 10 mg/L)
se acerca al extremo inferior del límite terapéutico (10 a 20 mg/L). En
algunos individuos, sobre todo en niños pequeños y recién nacidos sometidos a tratamiento anticonvulsivo, Km puede llegar a 1 mg/L. Cuando
la concentración deseada en un adulto es de 15 mg/L y ésta se alcanza
con una dosis de 300 mg/día, tomando en cuenta la ecuación 1-17, vm es
igual a 320 mg/día. Para estos pacientes, una dosis 10% menor que la óptima (esto es, 270 mg/día) producirá una Css de 5 mg/L, que es bastante
menor que el valor deseado. Por el contrario, una dosis 10% mayor que la
óptima (330 mg/día) excede a la capacidad metabólica (10 mg/día) y origina un ascenso lento y prolongado, pero constante, en la concentración
durante la cual aparecen efectos secundarios. La dosifi cación no se puede
regular con tanta precisión (error ,10%). Por lo tanto, para los pacientes
en quienes la concentración deseada de fenitoína es más de 10 veces
mayor que la Km, es casi imposible alternar entre un tratamiento ineficaz
y la aparición de efectos secundarios. Para un fármaco como fenitoína,
que posee un índice terapéutico estrecho y exhibe un metabolismo no
lineal, es muy importante determinar las concentraciones plasmáticas del
fármaco (véase más adelante en este capítulo). Cuando el paciente es un
neonato, este concepto cobra especial importancia puesto que los signos
y síntomas de los efectos adversos son difíciles de detectar. En estos casos, es recomendable consultar con un especialista en farmacocinética.
Diseño y optimización de los esquemas posológicos
Después de administrar un fármaco, sus efectos casi siempre exhiben un
patrón temporal característico (fig. 1-5). El efecto comienza posterior a
un periodo de retraso después del cual la magnitud del efecto aumenta hasta alcanzar el punto máximo y posteriormente desciende; si no
se administra otra dosis, el efecto termina por desaparecer conforme el
fármaco se elimina. Este fenómeno refl eja los cambios en la concentración del fármaco que dependen de la farmacocinética de su absorción,
distribución y eliminación. Por lo tanto, la intensidad de los efectos de
un fármaco depende de su concentración por arriba de la concentración
efectiva mínima, mientras que la duración del efecto refleja el tiempo
que la concentración permanece por arriba de esta cifra. En general, estas consideraciones se aplican para los efectos tanto deseados como indeseables (adversos) y, por lo tanto, existe una ventana terapéutica que
refleja el límite de concentración que es efectivo sin ocasionar efectos
secundarios. Lo mismo se observa después de administrar dosis múltiples en un tratamiento prolongado y define la cantidad de fármaco y la
frecuencia con que se debe administrar para lograr un efecto terapéutico
óptimo. En general, el límite inferior del rango terapéutico es similar a
la concentración del fármaco que produce la mitad del efecto terapéutico
mayor posible y el límite superior del rango terapéutico es tal que menos
de 5 a 10% de los pacientes experimenta efectos tóxicos. Para algunos
fármacos, esto signifi ca que el límite superior del rango no debe ser más
del doble del límite inferior. Por supuesto, estas cifras son variables y en
algunos pacientes se obtienen más benefi cios si se utiliza una concentración mayor que el límite terapéutico, mientras que otros padecen efectos
secundarios con una concentración mucho menor (p. ej., digoxina).
En lo que toca a un número escaso de fármacos, es posible medir con
facilidad parte del efecto (p. ej., en la presión arterial o la glucemia) y
ello puede servir para mejorar la dosis con un plan empírico de “tanteo”.
Incluso en esta situación ideal surgen problemas cuantitativos, como la
frecuencia con que debe cambiarse la dosifi cación, y el grado de estas
modificaciones; los dilemas mencionados pueden superarse con el uso
de reglas empíricas sencillas, basadas en los principios expuestos (p.
ej., no cambiar la dosifi cación más de 50%, ni con una frecuencia que
exceda de cada tres o cuatro semividas). Como otra posibilidad, algunos
agentes tienen una débil relación entre dosis y toxicidad, y casi siempre
se desea de ellos una máxima efi cacia. En estos casos, dosis mucho mayores que las promedio necesarias asegurarán la efi cacia (si es posible)
y prolongarán la acción farmacológica. Dicho plan de “dosis máxima”
se utiliza de manera clásica con las penicilinas.
Sin embargo, en muchos fármacos es difícil medir los efectos del medicamento (o éste se administra con fines profilácticos), hay el peligro latente de toxicidad e inefi cacia, o el índice terapéutico es muy estrecho. En
estas circunstancias, es necesario ajustar con enorme cuidado las dosis,
y éstas son limitadas por los efectos tóxicos más que por la eficacia. Sobre
tal base, el objetivo terapéutico es conservar valores de estado estable del
fármaco dentro de la ventana terapéutica. En lo que toca a casi todos los
medicamentos, no se conocen, ni es necesario identificar, las concentraciones reales que corresponden a estos límites deseados. Basta saber que
la efi cacia y los efectos tóxicos por lo común dependen de las concentraciones y la forma en que las dosis y la frecuencia de administración modifican el grado del efecto medicamentoso. Sin embargo, en un reducido
número de fármacos existe una diferencia pequeña (dos a tres tantos)
entre las concentraciones que producen efi cacia, y los efectos tóxicos
(como en el caso de la digoxina, la teofilina, la lidocaína, los aminoglucó-
sidos, la ciclosporina, la warfarina y los anticonvulsivos), y para dichos
fármacos se han defi nido límites de concentraciones plasmáticas que se
acompañan de efi cacia del tratamiento. En estos casos es razonable un
plan de “nivel preelegido”, es decir, en que casi siempre se selecciona
una concentración de estado estable elegida (meta) del medicamento (por
lo común en el plasma) que se acompaña de efi cacia y de efectos tóxicos
mínimos, y se calcula una dosis que a juicio del operador logrará dicho
objetivo. Más tarde se miden las concentraciones del fármaco y se ajusta
la dosifi cación si es necesario, para aproximarse en lo posible a la concentración deseada (véase más adelante en este capítulo).
Dosis de sostén. En seres humanos, los medicamentos casi siempre se
administran en una serie de dosis repetidas o por medio de goteo intravenoso continuo para conservar una concentración equilibrada y estable
vinculada con la ventana terapéutica. De este modo, el objetivo fundamental es calcular la dosis adecuada de sostén. Para conservar la concentración deseada o de estado estable, se ajusta el ritmo de administración
de modo que la velocidad de ingreso sea igual a la de egreso o pérdida.
Dicha relación fue definida en las ecuaciones 1-2 y 1-16, y se expresa en
este párrafo desde la perspectiva de la concentración deseada:
Frecuencia de dosificación 5 concentración deseada C
p . CL/F (1-18)
Si el clínico elige la concentración deseada del fármaco en plasma y
conoce sus cifras de eliminación y disponibilidad en un paciente particular, podrá calcular la dosis y el intervalo entre una y otra.
Ejemplo. Se va a administrar digoxina por vía oral en dosis de mantenimiento para “digitalizar” gradualmente a un paciente de 69 kg de
peso con insufi ciencia cardíaca congestiva. El objetivo es lograr una
concentración plasmática constante de 1.5 ng/ml, con base en lo que se
sabe sobre la acción de este fármaco en los pacientes con insuficiencia
cardíaca. Basados en el hecho de que la eliminación de creatinina del
paciente (creatinine clearance, CLCr) es de 100 ml/min, la eliminación
de digoxina se calcula tomando los datos del Apéndice II.



después de administrar la dosis del medicamento. Obtener especímenes durante la supuesta etapa de estado estable es modifi car el cálculo
de CL/F y, con ello, la selección de la posología. Las concentraciones
poco después de la absorción no expresan la eliminación; dependen más
bien de la tasa de absorción, el volumen de distribución “central” (no
el de estado estable) y la tasa de distribución, todas ellas características farmacocinéticas de muy poca importancia en la elección de dosis
de sostén para programas a largo plazo. Cuando la medición tiene por
objeto hacer ajustes en las dosis, la muestra debe obtenerse después de
dar la dosis previa (como regla empírica, apenas antes de la siguiente
dosis planeada, en que la concentración llega a su mínimo). Existe una
excepción: algunos fármacos son eliminados casi por completo entre
una y otra dosis, y actúan sólo en el lapso inicial del intervalo entre ellas.
En ese caso, si hay duda de que se estén alcanzando o no concentraciones efi caces, será conveniente obtener un espécimen poco después
de aplicar una dosis. Por otra parte, un motivo de preocupación es que
la poca eliminación, como en el caso de la insufi ciencia renal, puede
hacer que el medicamento se acumule, de modo que las concentraciones
medidas poco antes de la dosis siguiente indicarán si ha ocurrido dicha
acumulación, y los datos resultan mucho más útiles para ese fin que
conocer la concentración máxima. En tales situaciones, se recomienda
medir las concentraciones máxima y mínima. Estas dos cifras ofrecen
un panorama más amplio sobre la conducta del fármaco en determinado
paciente (en especial si se obtienen durante varios periodos posológicos) y corroboran mejor el modelo farmacocinético.
Un segundo aspecto importante al momento de obtener la muestra
es su relación con el comienzo del programa a base de dosis de sostén.
Si se da una dosis constante, el estado estable se alcanza sólo después
de que han transcurrido cuatro semividas. Cuando la muestra se obtiene
demasiado pronto después de iniciar el plan, no expresará con precisión
dicho estado ni la eliminación del fármaco. Sin embargo, en el caso de
medicamentos tóxicos, si se difi ere la obtención de especímenes hasta
que exista con seguridad un estado estable, para entonces muy probablemente ya habrá ocurrido daño. En estos casos pueden seguirse algunas pautas sencillas. Si se lleva a cabo un control cuidadoso de las concentraciones, se obtendrá el primer espécimen después de transcurridas
dos semividas (según se haya calculado y previsto para el paciente),
suponiendo que no se haya administrado dosis de saturación. Cuando la
concentración excede de 90% de la concentración media final esperada
de estado estable, habrá que dividir a la mitad la dosificación, obtener
otra muestra después de transcurridas otras dos semividas (supuestas)
y de nuevo disminuir a la mitad la dosifi cación si en la última muestra
el fármaco rebasa la cifra deseada. Si la primera concentración no es
demasiado alta, puede conservarse la dosifi cación inicial; aun cuando
la concentración sea menor que la esperada, el médico por lo común
espera que se alcance el estado estable después de otras dos semividas
calculadas, luego de lo cual ajusta la dosis como se describió antes en
este capítulo.
Cuando la dosifi cación es intermitente, surge un tercer problema
relacionado con el momento en que se obtuvieron las muestras para medir la concentración del medicamento. Si el espécimen se obtuvo poco
antes de la dosis siguiente, como se ha recomendado, la concentración
será la mínima y no la media. Sin embargo, como se mencionó antes,
es posible calcular la concentración media calculada por medio de la
ecuación 1-16.
Si el fármaco tiene cinética de primer orden, las concentraciones
promedio, mínima y máxima en estado estable guardan relación lineal con la dosis y con la dosificación (véanse ecuaciones 1-16, 1-19 y
1-20). Por tanto, para ajustar la dosis puede utilizarse la razón o cociente
entre las concentraciones medidas y las buscadas, congruente con la
magnitud de las dosis disponibles:
En el paciente del ejemplo descrito que recibía 0.375 mg de digoxina cada 24 h, por ejemplo, si se advertía que la concentración medida en
estado estable era de 1.65 ng/ml y no el valor buscado de 1.3 ng/ml, una
medida práctica y adecuada en cuanto al programa posológico hubiera
sido disminuir la dosis diaria a 0.25 mg de digoxina.
Observancia terapéutica
Por último, los buenos resultados con una terapéutica dependen de que
la persona cumpla fi elmente el plan medicamentoso, según lo haya
prescrito el médico (“los medicamentos no actúan si el sujeto no los
ingiere o los recibe por otras vías”). La falta de adherencia al esquema
posológico suele ser la razón principal de inefi cacia terapéutica, particularmente en el tratamiento a largo plazo con productos como los
antihipertensores, los antirretrovíricos y los anticonvulsivos. Cuando no
se hacen intentos especiales de atender este problema, sólo 50% de los
pacientes, en promedio, cumplirá con el plan recetado, de una forma
razonablemente satisfactoria; en promedio 33% mostrará observancia
parcial y uno de cada seis enfermos, aproximadamente, no cumplirá
en absoluto las órdenes recibidas. Es más frecuente el problema de las
dosis faltantes o no administradas que el de las “dosis excesivas”. El
número de fármacos al parecer no constituye un factor tan importante
como el número de veces al día que la persona debe recordar las dosis
(Farmer, 1999). La disminución de la cantidad de veces en que debe administrarse un medicamento mejorará el cumplimiento de un plan prescrito. De igual importancia es la necesidad de motivar a los pacientes y
hacer que participen en su propio cuidado, y para ello utilizar estrategias
diversas basadas en una mejor comunicación en cuanto a la naturaleza
de la enfermedad y al plan terapéutico global (véase Apéndice I).